
摘要
临床特征。
黏 脂贮积症IIIα/β (MLIIIα/β;假性胡勒多发性骨发育不良)是一种慢性进展性疾病,临床起病年龄约3岁,其特征是生长速度缓慢,矮身材;X线片显示轻度或中度多发性 骨发育不良;关节僵硬和疼痛,最初在肩关节、髋关节和指关节;颜面逐渐粗糙;认知正常或轻度受损。肝脾肿大一般较轻。儿童 期临床和放射学上明显的骨质疏松症的疼痛到青春期变得更加严重。心肺并发症(限制性肺部疾病,二尖瓣和主动脉瓣增厚和功能不全,左心室肥大和/或右心室 肥大)是导致成年早期或中期死亡的常见原因。
诊断/检测。
在MLIIIα/β中,由于对溶酶体水解酶缺乏靶向性,血浆或其他体液中几乎所有溶酶体水解酶的活性比正常对照高10倍。尿液中过量的寡糖(OSs)排泄是非特异性表现。GNPTAB编码的溶酶体水解酶N-乙酰葡糖胺-1-磷酸转移酶(GNPTA):该病UDP-N-乙酰葡糖胺-1-磷酸转移酶的活性显著不足(正常的1%-10%)。GNPTAB编码区域的双向测序能在95%以上的患者检出两种致病性变异。
处理。
治疗表现: 低强度物理疗法通常耐受性好。复发性中耳炎可以采取鼓膜切开置管术治疗。腕管综合征需要肌腱松解术来缓解。在儿童晚期或青春期早期,髋关节疼痛 症状初期可以用非处方镇痛药来缓解;一些症状较轻的青年、成年人,双侧髋关节置换很成功。在疾病后程,处理的重点是 缓解与骨质疏松相关的全身疼痛,部分患者对静脉注射双膦酸帕米膦酸盐有反应。
继发并发症的预防:由于气道管理方面的一些问题,外科手术仅应在有儿科麻醉师和重症监护医师的三级医疗机构进行。
随访:幼儿每年两次门诊随访;六岁以上儿童每年常规随访一次,骨痛、行走、心脏呼吸恶化时需要更加频繁监测。
遗传咨询。
MLIIIα/β 以常染色体隐性的方式遗传。患者同胞有25%的几率有可能受累,有50%的几率为无症状携带者,25%的几率既不受累,也不是携带者。如果家族中的致病性变异已知,可以进行高危亲属携带者测试和高危妊娠产前诊断。
诊断
临床诊断
以下临床表现有助于早期诊断黏脂贮积症IIIα/β型(MLIIIα/β)[Cathey 等人 2010],但不是诊断:
特殊表现:三岁出现 (婴儿晚期或儿童晚期)
生长缓慢
反复上呼吸道感染、中耳炎(易变)
关节僵硬最初在肩关节、髋关节和指关节
剧烈运动或物理治疗使关节疼痛加剧
颜面逐渐粗糙
裂隙灯检查可见角膜轻微混浊(易变)
肝脾肿大一般较轻
轻或中度不规则脊柱后突
认知正常或轻度受损
骨质疏松引起骨痛;儿童有临床和放射学表现,大龄患儿步态和关节运动范围受限
婴儿期和儿童早期骨骼X线片显示轻度或中度多发性 骨发育不良 [Spranger 等人 2002]:
长骨。起初正常或呈管状;股骨近端骨骺中度或重度发育不良
四肢。掌骨和指骨骨干轻度缩短;腕骨小
肋骨。正侧面(肋软骨交界处)较宽,但背面较狭窄
脊柱。轻度变扁;上、下终板不规则,背面呈扇形;下段胸椎或上段腰椎出现前下钩;椎间隙狭窄
骨盆。髂骨发育不良;髂骨翼外展;耻骨和坐骨延长;髋臼浅;髋外翻
颅骨。与身材相称;蝶鞍正常
童年晚期或青春期骨骼X线片显示:
长骨。股骨近端骨骺严重发育不良;整个骨骺消失。
手。小关节周围软组织硬化所致的手指爪状变形进展缓慢,骨骼影像学轻微异常。
脊柱。情况恶化;僵化;严重骨质减少的椎体形状显著变化;少数受累个体有严重的脊柱后突。
颅骨。颅骨逐渐增厚;蝶鞍少有延伸。
骨密度。骨质逐渐减少。
骨龄。手腕和长骨骨骺的骨化明显延迟。
检测
生物化学检测
溶酶体水解酶活性。MLIIIα/β患者血浆和其他体液中溶酶体水解酶活性几乎比正常对照高了10倍,由于甘露糖-6-磷酸(M6P)是溶酶体水解酶至溶酶体所必需的 ,缺乏M6P因而水解酶活性增加(参见分子遗传学发病机制)。
以下溶酶体水解酶最有意义,它们增加的活性与MLIII和溶酶体储存障碍鉴别诊断相关:
β-D-氨基己糖苷 (EC 3.2.1.52)
β-D-葡萄糖醛酸酶 (EC 3.2.1.31)
β-D-半乳糖苷酶 (EC 3.2.1.23)
α-D-甘露糖苷酶 (EC 3.2.1.24)
注意:MLIIIα/β患者酸性水解酶在白细胞内减少数量不定。与单一溶酶体酶缺乏引起的贮积障碍相反,MLIIIα/β不能通过测定白细胞中的酸水解酶来诊断。
寡糖尿液排泄(OSs)。这是一个简单而便宜的检测。过量OS是一种非特异性发现,临床误诊为寡糖贮积症。ML IIIα/β患者OS是易变的;假阴性结果很少见。
糖胺聚糖尿液排泄(GAG)(即酸性粘多糖[AMPS])是正常的。用以区分MLIII α/β和婴儿期后起病的MPS病。
UDP-N-乙酰氨基葡萄糖:溶酶体水解酶N-乙酰葡糖胺-1-磷酸转移酶(GNPTAB)酶活性。通过GNPTAB编码的UDP-N-乙酰氨基葡萄糖:溶酶体水解酶N-乙酰氨基葡萄糖-1-磷酸转移酶(GNPTA)(EC 2.7.8.17)在成纤维细胞显著缺失(正常的1%-10%),诊断MLIIIα/β[Kudo 等人 2005, Kudo 等人 2006]。GNPTAB酶活性的检测非常规,仅为临床诊断评估的一部分。
分子遗传学检测
基因。GNPTAB是已知致病性变异导致MLIIIα/β的唯一基因。
临床检测
表 1.
黏脂贮积症IIIα / β分子遗传学检测总结
| 基因1 | 检测方法 | 检测到致病性变异 2 | 变异检测频率 3 |
|---|---|---|---|
| GNPTAB | 序列分析 4 | 序列变体 | >95% 5 |
| 缺失/复制 分析 6 | 部分或整体基因 缺失或复制 | 未知;未报道 7 |
- 1.
见染色体 位点和蛋白质的表 A. 基因和数据库
- 2.
见分子遗传学等位基因变异信息
- 3.
用于检测指定基因中变异的测试方法。
4. 序列分析检测是好的,可能是好的,不确定意义的,可能是致病的或致病的。致病性变异可能包括小基因缺失/插入和错义、无义、剪接位点变异;通常,未检测到外显子或全基因缺失/重复。要解决序列分析结果问题,请点击此处。
- 5.
超过95%的MLIIIα/β患者整个GNPTAB编码区双向测序检测出两种致病性变异。
- 6.
鉴定基因组DNA编码和侧翼内含子区域的序列分析实验不容易检测到缺失/重复;可以使用多种方法包括:定量PCR,长片段PCR,多重连接依赖性探针扩增(MLPA)和包括该基因/染色体片段的染色体微阵列(CMA)。
- 7.
变异检测率未知,可能非常低。
检测结果说明。如果单个致病性等位基因变异为明显纯合性,检测每个亲本的携带者状态以确定其孩子是否是缺失和变异的复合杂合子。
检测方法
确认/建立先证者的诊断需要兼顾临床评估和实验室检测。推荐以下检测顺序:
- 1.
识别特征性临床和影像学检查结果
- 2.
尿中寡糖(OS)的测定
- 3.
血浆中几种酸性水解酶的测定;例如:
β-D-氨基己糖苷 (EC 3.2.1.52)
β-D-葡萄糖醛酸酶 (EC 3.2.1.31)
β-D-半乳糖苷酶 (EC 3.2.1.23)
α-D-甘露糖苷酶(EC 3.2.1.24)
芳基磺胺酶A (EC 3.1.6.1)
注意:(1)MLIII患者血浆中溶酶体酶的比活性升高,成纤维细胞中降低,白细胞中正常。(2)溶酶体水解酶在白细胞中的比活性可用于和其他晚期发病溶酶体病症的鉴别诊断,但在MLIIIα/β本身的诊断中没有价值。
- 4.
GNPTAB的序列分析。
- 5.
GNPTAB的缺失/复制分析;适用于:
危险亲属的携带者检测依赖于分子基因检测。优选先前鉴定的家族致病性变异;如果受累儿童无法检测,则可以进行携带者双亲全基因 序列分析,以鉴定两种致病性等位基因。
注意:酶活性的评估无法可靠地鉴定杂合的个体。
预测。分子遗传学研究的基因型 - 表型相关性支持ML III α/β和ML II(等位基因,临床上表现更严重)的临床区别。完全灭活磷酸转移酶原始酶的致病性变异导致ML II,不管其在基因内的位置如何。对这种酶活性有鲜有影响的致病性变异通常导致ML IIIα/β或罕见的中间表型。[Paik 等人 2005, Steet 等人 2005, Tiede 等人 2005, Bargal 等人 2006, Kudo 等人 2006, Otomo 等人 2009, Tappino 等人 2009, Cathey 等人 2010, David-Vizcarra 等人 2010].
产前诊断和植入前遗传学诊断(PGD)对于高危妊娠,需事先鉴定家族中的致病性变异。
临床特征
临床描述
黏脂贮积症IIIα/β(MLIIIα/β;假性胡勒多发性骨发育不良)是一种慢性进展性先天代谢异常疾病,临床起病年龄约3岁,致命表现出现在成年早期或中期[Leroy 2007, Cathey 等人 2010]。目前仍缺乏关于预期寿命的综合数据。
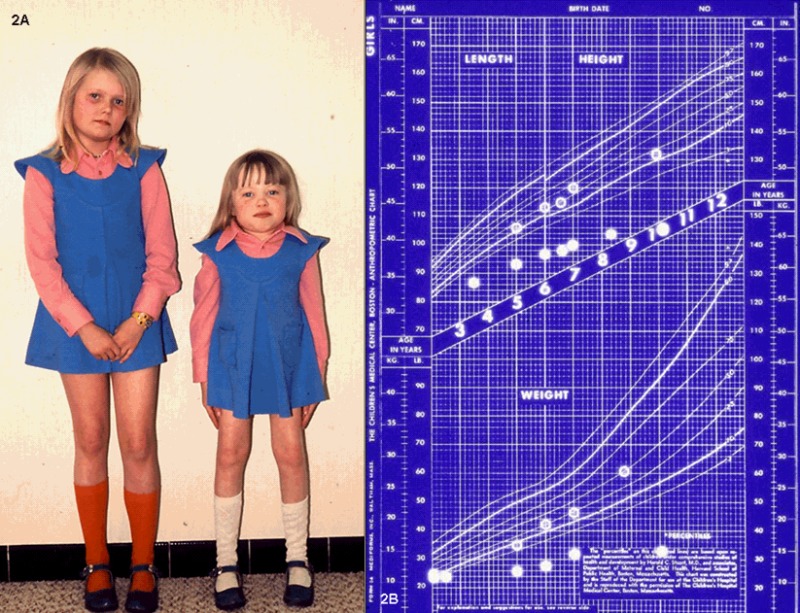
生长。出生时的体重和身高在正常范围内。从婴儿期晚期至幼儿期,增长速度逐渐放缓。三岁以前很少出现身材矮小的问题,此后肩关节、髋关节和膝盖关节挛缩,加重了对身高的不利影响。ML IIIα/β不像ML II那样引起弗兰克侏儒症(见图 1);但从儿童早期开始,患儿的身高往往低于标准生长曲线的第三个百分位点(见图 2A,图 2B)。最终身高远低于预期。
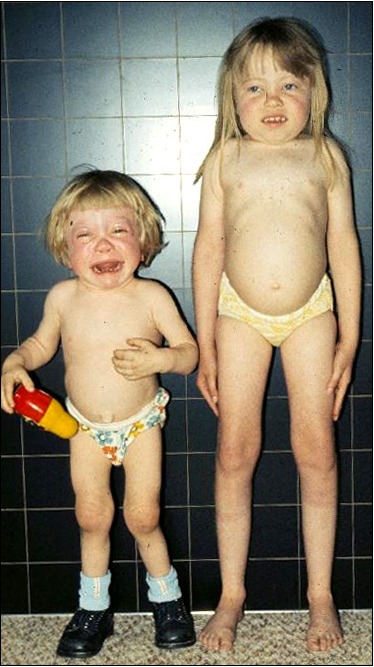
颅面。无巨头畸形。幼童没有或少有面部畸形。颜面逐渐粗糙,包括丰满的脸颊,凹陷的鼻梁和突出的嘴巴。牙龈肥大是轻度的,通常不会影响牙齿萌出。
眼科。内眦赘皮持续的时间延长。眼球突出(ML II经常能观察到)少见。角膜通过常规检查可以看清,但角膜浑浊只有在裂隙灯下才能看到。
听力。ML IIIα/β患者较正常人发生中耳炎的频率更高。尚未有系统研究记录受累患者听力损伤。感觉神经性听力损伤不是ML III的典型特征。
呼吸。声音反复嘶哑。一些(但不是全部)儿童频繁患上呼吸道感染。从儿童晚期开始,支气管炎和支气管肺炎是最持续的临床并发症。
成人表现出胸壁僵硬引起的肺部疾病、支气管慢性进行性硬化,以及肺实质内间质组织(细胞外基质)的硬化和增厚。
心血管。MLIIIα/β患者有心脏受累的风险。二尖瓣和主动脉瓣逐渐增厚及瓣膜不全常见于儿童后期[Steet 等人 2005]。
MLIIIα/β患者少有快速进展的心脏病。
肺炎可能合并轻度心功能不全。成年早期的死亡原因往往是肺心病。
胃肠道。腰椎间盘突出症、髋关节和膝关节屈曲挛缩以及腹壁肌肉肌张力减低导致直立时腹部突出。可能存在内侧直肠疝和脐疝。通常MLIIIα/β患者没有器官肿大。
骨骼/结缔组织。所有大小关节僵硬是一个主要特征。结缔组织病变导致肩关节活动范围受限常常是ML IIIα/β的初步证据。
髋关节和膝关节运动范围受限导致儿童步行困难,无法跑步。 髋关节和膝关节的屈曲挛缩会引起蹲姿,晚期最为明显(见图 3)。
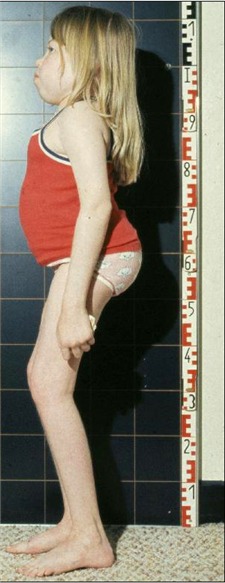
髋关节严重的关节炎可导致股骨近端骨折破坏,使步行越来越困难痛苦。髋关节周围软组织的显著僵硬加剧了髋关节功能障碍。许多受累患者在成年早期需要使用轮椅。
手腕和脚踝的运动范围轻度受限。儿童晚期可能出现Dupuytren型手掌挛缩,手指反复肿胀僵硬,呈中度至重度爪状畸形。一些患者腕管综合征的神经症状加重。
ML IIIα/β患者手和手指长度近似正常,与ML II严重受累相反。
在确诊之前,许多MLIIIα/β患者曾被诊断为风湿病。
骨质疏松影响骨骼。骨骼疼痛是ML IIIα/β中最痛苦的症状。溶骨损伤也与患者剧烈骨痛卧床不动有关。
神经运动和智力发育是ML IIIα/β中最易变的表现,运动发育轻度或中度延迟。接受性和表达性语言发育正常。ML IIIα/β患者中没有观察到口吃。虽然心理测试显示智力正常,但大约一半受累儿童常因为身体的缺陷而需要学校帮助。
其他
颈短。
皮肤增厚不均匀。
以前使用的诊断检测。过去通过相差显微镜或电子显微镜(EM)观察成纤维细胞中大量致密细胞质包涵体(I细胞),来证实ML II和MLIIIα/β的诊断(参见图 4)。
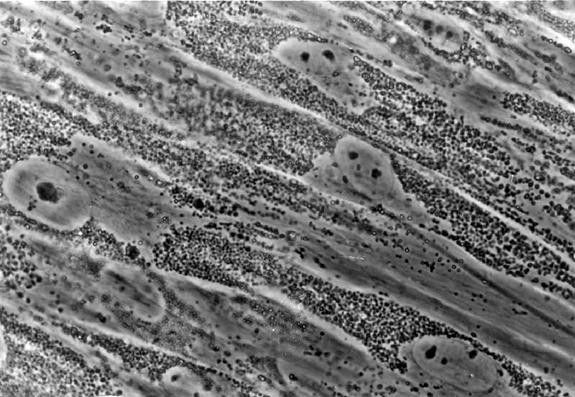
注意:在电子显微镜(EM)中,任何组织中的间充质细胞包含大量细胞质空泡(肿胀的溶酶体),呈多形,但不密集。这是ML II和MLIIIα/β特有的,在任何其他溶酶体储存障碍中都没有观察到。
溶酶体酶的活性在I细胞中严重降低,但在相应的培养基中显着增加。
细胞学和细胞培养酶学无法区分ML II(I细胞病)和ML IIIα/β(参见ML II)。
基因型-表型相关性
2005年之后对GNPTAB进行测序;几项研究结果表明,没有或几乎没有GlcNAc-1磷酸转移酶(由过早翻译终止或移码效应引起)的纯合和复合杂合基因型导致ML II表型。 较少的“病态”致病性变异(例如错义和大多数导致多达10%残留GlcNAc-1 - 磷酸转移酶活性的剪接位点变异)的组合通常导致MLIIIα/β表型 [Tiede 等人 2005, Paik 等人 2005, Bargal 等人 2006, Kudo 等人 2006, Encarnaçao 等人 2009, Otomo 等人 2009, Tappino 等人 2009, Cathey 等人 2010, David-Vizcarra 等人 2010, Cury 等人 2011]。
一些儿童临床表型之间的中间表型,参考已描述的ML II和MLIIIα/β表型[Cathey 等人 2010, David-Vizcarra 等人 2010]。经验丰富的临床医生经常在这些ML II / III患者中标记基于GNPTAB突变的疾病。一些中间表型与特异性突变基因型一致相关,而其他的中间表型有纯合突变基因型。
目前尚不知道和ML II类似研究相比,ML IIIα/β[Kerr 等人 2011, Kobayashi 等人 2011]中极少可用于死后病理学研究是否会增加基因型 - 表型相关性。这些研究显然有有助于发现发病机制。
命名法
假性胡勒多发性骨发育不良(PHP)是Maroteaux和Lamy在1966年使用的术语,他们首先在临床和影像学上发现与Hurler病或黏多糖贮积症I型(MPS I)相同的大小关节进行性僵硬这一多系统疾病。
黏脂贮积症(ML)。术语PHP在很大程度上取代了1970年 由Spranger和Wiedemann提出的黏脂贮积症Ⅲ型,这些术语提供了临床上介于脂质沉积和MPSs(黏多糖贮积症)之间代谢紊乱组的第一个临 床分类。他们的假说认为,这些疾病可能是遗传相关的,1973年通过“体外”细胞学以及在PHP患者成纤维细胞内发现的I 细胞现象解释了ML III(PHP)和ML II(I细胞疾病)。
虽然黏脂贮积症这一名称在临床上使用,但生化学家认为它是一种误称,因为“黏脂”本身并不存在。术语黏脂贮积症已被用于四种不同的先天性代谢疾病;只有ML II和ML IIIα/β与GNPTAB相关。黏脂贮积症I型(也称为涎酸贮积症II型)和黏脂贮积症IV型是遗传上明显不同的疾病。
寡糖贮积病(OSs)。20世纪70年代,大多数黏脂贮积症记录到过量排泄的OSs;因此,术语“寡糖贮积病”和后来的术语“糖蛋白贮积病”已被术语黏脂贮积症替代。
黏脂贮积症 II型、黏脂贮积症IIIα/β型和黏脂贮积症 IIIγ型。UDPGlcNAc-磷酸转移酶的致病性酶缺陷名称很长,目前ML II和ML III α /β的命名为UDP-GlcNAc 1-P转移酶缺陷障碍,严格上说,这个命名才是最正确的。[Leroy 2007]
GNPT酶是两个基因的产物,一个编码α和β亚基,另一个编码γ亚基[Bao 等人 1996]:
GNPTAB中的致病性变异导致MLIIIα/β和等位基因失调ML II。
GNPTG中的致病性变异导致ML III型,称为MLIIIγ [Cathey 等人 2008]。
流行病学
根据客观数据无法估计ML IIIα/ β的流行率。然而,有可能流行率与ML II的患病率相同,估计在2.5×10-6至1.10-5之间。
携带者概率估计在1:158至1:316之间(见ML II)。
遗传相关(等位基因)疾病
黏脂贮积症II型(ML II,I型细胞病)和MLIIIα/β是等位基因。ML II出生即发病。ML II的进展比ML III更严重,预期寿命显著缩短。已经观察到ML II和MLIIIα/β中间表型相关的基因型。在大多数中间表型复合杂合子中,观察到了致病性错义和剪接位点变异。
GNPTAB中的致病性变异引起的疾病临床表型分为两种[Cathey 等人 2010],非“经典”型ML II和MLIIIα/β之间的连续变异谱。在分子分析之前[Paik 等人 2005, Steet 等人 2005, Tiede 等人 2005, Bargal 等人 2006, Kudo 等人 2006, Cathey 等人 2008, Encarnaçao 等人 2009, Otomo 等人 2009, Tappino 等人 2009], 从ML IIIα/β中划分ML II完全取决于临床标准,包括发病年龄、进展速度和总体严重程度。
注意:黏脂贮积症 III γ,虽然临床上与MLIIIα/β类似,但不是等位基因疾病。见分子遗传学。
鉴别诊断
黏脂贮积症 III γ 。黏脂贮积症III型γ(也称为变异的ML III)临床特征与在黏脂贮积症IIIα/β(MLIIIα/β)患者中观察到的相似,但程度较轻。 在大多数已发表的ML IIIγ病例报告中,受累患者都是中东血统 [Raas-Rothschild 等人 2000, Raas-Rothschild 等人 2004, Cathey 等人 2008]。如果强烈怀疑ML III的诊断,并且分子分析显示GNPTAB非致病变体,则应进行GNPTG分析。
见 命名法。
溶酶体贮积病。ML IIIα /β患者的临床表现与MPSs中几乎所有迟发性轻型描述性实体观察到的结果重叠,包括:
轻型 MPS I (原先称为Hurler-Scheie综合征或Scheie综合征)
轻型 MPS II (Hunter综合征)
Morquio综合征B型 (MPS IV B)
Maroteaux-Lamy综合征B型 (MPS VI B)
Sly综合征B型 (MPS VII B)
多发性骨发育不良实体与体格检查有更严重的贮积相关证据。MPSs患儿头部增大,这一发现不存在于ML III α/β中。生化测试区分MPSs。
在一组OSs中,更具挑战性的鉴别诊断包括:α-甘露糖苷症,婴儿晚期和青少年半乳糖涎酸贮积症,以及儿童期异形的涎酸贮积症 (ML I) [Leroy 2007]。
寡糖贮积症。临床上与鉴别诊断相关的寡糖贮积症包括婴儿晚期涎酸贮积症或Salla病(见游离涎酸贮积症)和多种硫酸酯酶缺乏症(粘硫脂病)。在这两种疾病中,神经变性更严重。在游离涎酸贮积症中,不存在多发性骨发育不良或最小尿游离涎酸(不是OS)排泄过多。在多种硫酸酯酶缺乏症中,尿中的AMPS和硫苷脂都过量。
风湿性疾病。由于大、小关节运动范围缓慢受限,髋部疼痛加重,ML IIIα/β患者常被怀疑为风湿性疾病。类风湿性关节炎临床和实验室检查显示有炎症和特异性抗体;白细胞中溶酶体酶的活性是正常的。不存在多发性骨发育不良。
骨软骨发育不良与MLIIIα/β具有临床相似性但没有多发性骨发育不良的放射学特征[Spranger 等人 2002]包括以下内容:
常染色体显性早熟性骨关节病,迟发型II型胶原基因病;遗传异质性
进行性类风湿性软骨发育不良;常染色体隐性
多发性骨骺发育不良 (见多发性骨骺发育不良,隐性和多发性骨骺发育不良,显性)
处理
初步诊断后评估
要确诊黏脂贮积症IIIα/β(ML IIIα/β )患者疾病程度,建议进行以下评估:
如果没有进行诊断性评估或诊断性评估不完整,需进行放射学检查。
请骨科医生和代谢骨科专家进行基线评估,以便更好地确定是否或何时可以启动手术干预或二磷酸盐治疗 (见临床表现的治疗)
心脏超声心动图评价瓣膜增厚及心室大小和功能
眼科检查
听力筛查
发育评估,帮助建立对儿童生长发育的适当期望
遗传咨询
临床表现的治疗
指导支持治疗和对症治疗。
骨骼。对大、小关节运动的进行性范围受限无有效的治疗。经典的物理治疗早期干预项目通常对发育迟缓、神经运动迟缓或脑瘫儿童有益,不能明确地推荐给ML IIIα/β患儿,原因如下:
拉伸练习无效且痛苦。
没有经验的治疗师可能会伤害周围的关节囊和相邻的肌腱,导致随后的软组织钙化。
对于关节和肌腱劳损的“低强度”治疗,包括短程水疗法,通常有很好的耐受性。
儿童晚期或青春早期注意步行和髋部疼痛的治疗需要。
腕管综合征,需行肌腱松解手术,暂时缓解症状。
病程后期,骨痛程度易变。
几位ML IIIα/β患者已经获得了令人满意的结果,每月静脉注射给予帕米磷酸盐(一种双磷酸盐)。推荐剂量为每月1 mg / kg。正在开发的方案与适用于成骨不全症的不同个体。需要密切监测骨密度。目前,在疾病进程中何时使用或几岁开始这种治疗的资料不足。在开始治疗的几个月内,已经公开信息的两位患者,骨痛减轻了。一些曾使用轮椅的患者,恢复行动已经一年以上。骨密度测定法已得到了改进[Robinson 等人 2002]。
需要就此对症治疗做几点评价:
父母和受累患者必须记住,这种治疗不能治愈疾病。它既不能抑制骨质吸收也不会改变其过程。
长期的影响是未知的。
治疗方案的终点不确定[Robinson 等人 2002; Sillence, 个人通信]。
受累患者均受益于双磷酸盐的治疗。在ML IIIα/β和其他骨病中使用双磷酸盐治疗是全球一个热点临床研究领域。
在患ML IIIα/β表型变异程度较轻的大龄青少年和成年人中,双侧髋关节置换已成功。
听力。复发性中耳炎在ML IIIα/β患者中经常发生。患病率随着年龄的增长而下降。鼓膜切开导管放置术被认为是传导性听力缺陷的必需预防措施,但由于患者特殊的气道问题和所涉及的麻醉风险(参见继发性并发症的预防),此手术不应被视为“常规”手术。
预防继发并发症
由于气道管理方面的一些问题,外科手术仅应在有儿科麻醉师和重症监护医师的三级医疗机构进行。ML IIIα/β患者身材矮小,气道狭窄,僵硬的结缔组织使气管柔韧性降低,粘膜增厚使管腔逐渐缩窄。使用与年龄和大小匹配的儿童气管内导管是必要 的。胸腔依从 性差及逐渐硬化的肺实质进一步使气道管理复杂化。必须使用纤维支气管插管。
胸廓依从性差及逐渐硬化的肺实质进一步使气道管理复杂化,特别是老年人。 肺实质的功能减退可能至少部分是由于结缔组织在细胞外基质中缓慢进行性退化,这是一个未充分研究但伴随骨质减少的现象。由于亚临床心脏衰竭可能在麻醉期 间出现,任何外科手术之前都应进行彻底的心脏评估。拔管也可能是一个挑战。
监测
ML IIIα/β患者每年两次门诊随访。
从六岁开始,每年随访,除非骨痛和行走障碍恶化,需要关注心脏及呼吸监测。
危险亲属评估
研究中的治疗方法
初步结果表明,静脉注射二磷酸盐可以减轻一些受影响个体的骨痛;然而,这种治疗仍被认为是研究性的。
搜索ClinicalTrials.gov获取关于各种疾病和病症的临床研究信息。
遗传咨询
遗传咨询是向个人和家庭提供关于遗传疾病的性质、继承和影响的信息,以帮助他们做出明智的医疗和个人决定的过程。以下部分涉及遗传风险评估和使 用家族史及遗传检测来澄清家庭成员的遗传状况。本节并不意味着解决个人面临的所有个人、文化或伦理问题,或代替咨询遗传学专业人士。—ED.
遗传模式
黏脂贮积症 IIIα/β(ML IIIα/β)以常染色体隐性遗传的方式遗传。
家庭成员的风险
先证者的父母
先证者的同胞
先证者的后代。ML IIIα/β患者通常无法生育。MLIIIα/β的患者后代是GNTAB中致病性变异的专性杂合子(携带者)。
先证者的其他家庭成员每一个先证者父母的同胞有50%的风险是携带者。
携带者(杂合子)检测
一旦家庭中发现了致病性变异,可以对高危家庭成员进行携带者检测。
相关遗传咨询问题
参见管理、评估风险亲属的信息,以便及早诊断和治疗高危亲属。
计划生育
DNA银行是DNA的存储库(通常从白细胞中提取),以备日后使用。因为测试方法和基因、等位基因变异及疾病的理解在将来会有所改善,所以考虑将受累患者的DNA储存起来。
产前检测及植入前遗传学诊断
一旦在受累家族成员中检测到GNPTAB 致病变体,就可以进行怀孕风险增加的产前诊断和植入前遗传学诊断。
资源
GeneReviews的工作人员选择了以下具体疾病和/或伞式支持组织和/或注册管理机构,以造福患有这种疾病的个人及其家属。GeneReviews不对其他组织提供的信息负责。 有关选择标准的信息,请点击此处 。
- International Advocate for Glycoprotein Storage Diseases (ISMRD)3921 Country Club DriveLakewood CA 90712Email: info@ismrd.org
- National MPS SocietyPO Box 14686Durham NC 27709-4686Phone: 877-677-1001 (toll-free); 919-806-0101Fax: 919-806-2055Email: info@mpssociety.org
- Society for Mucopolysaccharide Diseases (MPS)MPS House Repton PlaceWhite Lion RoadAmersham Buckinghamshire HP7 9LPUnited KingdomPhone: 0345 389 9901Email: mps@mpssociety.co.uk
分子遗传学
分子遗传学和OMIM表中的信息可能与GeneReview中的其他信息不同;表可能包含更多最新信息。—ED.
表 A.
黏脂贮积症IIIα/β:基因和数据库
| 基因 | 染色体位点 | 蛋白 | 位点特异性 | HGMD |
|---|---|---|---|---|
| GNPTAB | 12q23 | N-乙酰氨基葡萄糖-1-磷酸转移酶亚基α/β | GNPTAB 数据库 | GNPTAB |
数据来自以下标准参考文献: HGNC基因;染色体位点,基因座名称,临界区,OMIM互补基团; 来自UniProt的蛋白质。有关提供链接的数据库(Locus Specific,HGMD)的描述,请点击此处。
表 B.
OMIM 收录的黏脂贮积症IIIα/β (看OMIM内所有内容)
| 252600 | MUCOLIPIDOSIS III ALPHA/BETA |
| 607840 | N-ACETYLGLUCOSAMINE-1-PHOSPHOTRANSFERASE, ALPHA/BETA SUBUNITS; GNPTAB |
分子遗传学发病机制
UDP-N-乙酰葡糖胺的部分灭活:溶酶体水解酶N-乙酰葡糖胺1-磷酸转移酶(由GNPTAB和GNPTG编码[参见黏脂贮积症IIIγ])可由致病性变异导致蛋白质产生减少或残留(错义或 部分剪切位点变异)。因此,普通甘露糖-6-磷酸(M6P识别标记)部分对溶酶体酸水解酶的合成显著降低,但未完全消除。与高尔基体外侧网络中特异性 M6P受体的结合是非常不足的,并导致受体介导的溶酶体酶向溶酶体细胞内室的无效转运。突变的水解酶离开细胞并在体液以及体外细胞培养基中过量出 现。一旦出来,酶便不能重新进入正常成纤维细胞(因此通常被称为“低摄取”溶酶体酶)。相比之下,正常结构的磷酸糖蛋白(“高摄取”溶酶体酶)的正常成熟 酸水解酶可以通过胞饮作用进入任何类型培养的成纤维细胞(包括“I细胞”)[Kornfeld & Sly 2001]。MLIIIα/β患者和MLIIIγ患者的体液及组织细胞培养基中的低摄取酶之间的定量差异是微不足道的,体内细胞间基质中尚未研究。
溶酶体水解酶的N-连接糖基化发生在内质网中,它是先前OS逐步堆积的位点,它的“整体”从二氢乙酰基磷酰基-OS-前体载体转移到新生水解酶蛋白质中的一些天冬酰胺残基中。
随 着新形成的糖蛋白穿过高尔基体,N连接的OS顺序酶修饰沿着两个不同的途径发生:一个途径将N连接的OS修饰为复合型聚糖侧链,而另一个途径则是更重要的 途径,至少在间充质组织中,将前体聚糖定量地转化为寡聚半乳糖型OS侧链。单独的特异性磷酸化在该合成途径的晚期步骤中,由于双等位基因GNPTAB使致病变体失活,受到不利影响(见下一段)。这种显著降低的特异性磷酸化在临床上表现为MLIIIα/β;而完全缺乏寡聚半乳糖聚糖的磷酸化则导致ML II。在MLIIIα/β中,溶酶体水解酶内识别标记物M6P的形成显著降低,在ML II患者中几乎或完全不存在。
M6P识别标记的正常形成是两步法。第一步由UDP-N-乙酰葡糖胺催化:溶酶体水解酶N-乙酰葡糖胺-1-磷酸转移酶(简称GlcNAc-磷酸转移酶GNPTAB)(EC 2.7.8.17)催化。该酶也称为N-乙酰葡糖胺(GlcNAc)-1-磷酸转移酶,分别包含由GNPTAB编 码的天然蛋白质亚基α和β,分别为氨基和羧基末端。该酶的失活或缺乏分别导致ML II和MLIIIα/β。第二步涉及N-乙酰葡糖胺-1-磷酸二酯α-N-乙酰氨基葡萄糖苷酶的作用,在ML II或IIIα/β患者中不受影响,其从磷酸化中除去封闭的N-乙酰葡糖胺(GlcNAc)残基寡糖基型OR,从而暴露识别标记物M6P。迄今为止,在 ML II或MLIIIα/β中尚未报道使N-乙酰葡糖胺-1-磷酸二酯α-N-乙酰氨基葡糖苷酶失活的致病性变异。
基因结构。位于染色体12q23.3上的GNPTAB是一个有21个外显子并跨85kb的基因组DNA。GNPTAB在单个6.2-kb的α/β转录物中编码低聚的人类GNPTAB的α和β亚基(参见分子遗传学发病机制)。 参见表 A,基因,了解基因和蛋白质信息的详细总结。
致病性等位变异。已知有几十种致病性变异。类型包括:(a)错义,无义及剪接位点变体;和(b)小的插入和缺失,导致阅读框架的移动(见表 2 [pdf])。可以在MLII患者中找到与剪接位点或错义变异组合的任意一种类型变异。大多数MLIIIα/β患者是纯合子 或复合杂合体,由于错义或剪接位点变异。一些存活时间长、有轻微症状和体征的表型与检测到的突变基因相关型有关。[Paik 等人2005, Steet 等人 2005, Tiede 等人 2005, Bargal 等人 2006, Kudo 等人 2006, Encarnaçao 等人 2009, Otomo 等人 2009, Tappino 等人 2009, Cathey 等人 2010]。迄今为止,ML IIIα/β患者中没有报道更大规模的重排。
正常基因产物。GNPTAB将寡聚的人类GNPTABα和β亚基编码在单个6.2-kb的α/β转录物中。
人类GNPTAB酶的亚基结构是由三个亚基组成的540-kd六聚体复合物,这三个亚基是二硫键连接的同型二聚体。每个由1个166-kd的α亚基(由GNPTAB前体基因编码)和1个51-kd的γ亚基(由GNPTG编码)组成。这些亚复合物中的每一个与1个56-kdβ亚基(由GNPTAB前体基因编码)非共价结合。因此,六聚体酶复合物可以被表示为α2β2γ2 [Kudo 等人 2005, Tiede 等人 2005, Kudo 等人 2006]。
翻译后,α/β前体多肽在赖氨酸(残基928)- 天冬氨酸(残基929)肽键处进行蛋白水解切割。该键通过位点-1蛋白酶(S1P)酶促释放。S1P缺陷型细胞未能激活α/β前体并在体外显示I细胞表型[Marschner 等人 2011]。N-末端α亚基,其中较大的两个,由928个氨基酸组成。β亚基,前体的C-末端部分含有328个氨基酸。1256-氨基酸前体蛋白分子量预测为144kd,两个跨膜结构域和19个潜在的糖基化位点[Kudo 等人 2005, Tiede等人 2005, Kudo 等人 2006]。在最近对ML II和MLIIIα/β成纤维细胞(I细胞)菌株的研究中,使用针对α和β亚基的抗肽抗体显示,γ亚基中的致病性变异不利地影响前亚基的组装和细胞内分布[Zarghooni & Dittakavi 2009]。
参考文献
Literature Cited
- Bargal R, Zeigler M, Abu-Libdeh B, Zuri V, Mandel H, Ben Neriah Z, Stewart F, Elcioglu N, Hindi T, Le Merrer M, Bach G, Raas-Rothschild A. When mucolipidosis III meets mucolipidosis II: GNPTA gene mutations in 24 patients. Mol Genet Metab. 2006;88:359 - 63. [PubMed: 16630736]
- Bao M, Booth JL, Elmendorf BJ, Canfield WM. Bovine UDP-N-acetylglucosamine:lysosomal-enzyme N-acetylglucosamine-1-phosphotransferase. I. Purification and subunit structure. J Biol Chem. 1996;271:31437 - 45. [PubMed: 8940155]
- Cathey SS, Kudo M, Tiede S, Raas-Rothschild A, Braulke T, Beck M, Taylor HA, Canfield WM, Leroy JG, Neufeld EF, McKusick VA. Molecular order in mucolipidosis II and III nomenclature. Am J Med Genet. 2008;146A:512 - 3. [PubMed: 18203164]
- Cathey SS, Leroy JG, Wood T, Eaves K, Simensen RJ, Kudo M, Stevenson RE, Friez MJ. Phenotype and genotype in mucolipidoses II and III alpha/beta: a study of 61 probands. J Med Genet. 2010;47:38 - 48. [PMC free article: PMC3712854] [PubMed: 19617216]
- Cury GK, Velho RV, Alegra T, Matte U, Artigalas O, Burin M, Ribeiro EM, Lourencnullo CM, Kim CA, Valadares ER, Miguel DSCG, Acosta AX, Galera MF, Schwartz IVD. Mucolipidoses type II and III: molecular analysis of the GNPTAB and GNPTG genes in 15 Brazilian patients. J Inher Metab Dis. 2011;34:S206.
- David-Vizcarra G, Briody J, Ault J, Fietz M, Fletcher J, Savarirayan R, Wilson M, McGill J, Edwards M, Munns C, Alcausin M, Cathey S, Sillence D. The natural history and osteodystrophy of mucolipidosis types II and III. J Paediatr Child Health. 2010;46:316 - 22. [PMC free article: PMC4188554] [PubMed: 20367762]
- Encarnação M, Lacerda L, Costa R, Prata MJ, Coutinho MF, Ribeiro H, Lopes L, Pineda M, Ignatius J, Galvez H, Mustonen A, Vieira P, Lima MR, Alves S. Molecular analysis of the GNPTAB and GNPTG genes in 13 patients with mucolipidosis type II or type III - identification of eight novel mutations. Clin Genet. 2009;76:76 - 84. [PubMed: 19659762]
- Kerr DA, Memoli VA, Cathey SS, Harris BT. Mucolipidosis type III α/β: the first characterization of this rare disease by autopsy. Arch Pathol Lab Med. 2011;135:503 - 10. [PMC free article: PMC4188553] [PubMed: 21466370]
- Kobayashi H, Takahashi-Fujigasaki J, Fukuda T, Sakurai K, Shimada Y, Nomura K, Ariga M, Ohashi T, Eto Y, Otomo T, Sakai N, Ida H. Pathology of the first autopsy case diagnosed as mucolipidosis type III α/β suggesting autophagic dysfunction. Mol Genet Metab. 2011;102:170 - 5. [PubMed: 21051253]
- Kornfeld S, Sly WS. I-Cell disease and pseudo-Hurler polydystrophy: disorders of lysosomal enzyme phosphorylation and localization. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Vogelstein B, eds. The Metabolic and Molecular Bases of Inherited Disease. 8 ed. New York, NY: McGraw-Hill; 2001:3469-82.
- Kudo M, Bao M, D’Sousa A, Ying F, Pan H, Roe BA, Canfield WM. The α- and β-subunits of the human UDP-N-acetylglucosamine: lysosomal enzyme N-acetylglucosamine-1-phosphotransferase [corrected] are encoded by a single cDNA. J Biol Chem. 2005;280:36141 - 9. [PubMed: 16120602]
- Kudo M, Brem MS, Canfield WM. Mucolipidosis II (I-cell disease) and Mucolipidosis III (classical pseudo-Hurler polydystrophy) are caused by mutations in the GlcNAc-phosphotransferase alpha / beta -subunits precursor gene. Am J Hum Genet. 2006;78:451 - 63. [PMC free article: PMC1380288] [PubMed: 16465621]
- Leroy JG. Oligosaccharidoses, disorders allied to the oligosaccharides. In: Rimoin DL, Connor JM, Pyeritz RE, Korf BR, eds. Emery and Rimoin’s Principles and Practice of Medical Genetics. 5 ed. Philadelphia, PA: Churchill Livingstone; 2007:2413-48.
- Marschner K, Kollmann K, Schweizer M, Braulke T, Pohl S. A key enzyme in the biogenesis of lysosomes is a protease that regulates cholesterol metabolism. Science. 2011;333:87 - 90. [PubMed: 21719679]
- Otomo T, Muramatsu T, Yorifuji T, Okuyama T, Nakabayashi H, Fukao T, Ohura T, Yoshino M, Tanaka A, Okamoto N, Inui K, Ozono K, Sakai N. Mucolipidosis II and III alpha/beta: mutation analysis of 40 Japanese patients showed genotype-phenotype correlation. J Hum Genet. 2009;54:145 - 51. [PubMed: 19197337]
- Paik KH, Song SM, Ki CS, Yu HW, Kim JS, Min KH, Chang SH, Yoo EJ, Lee IJ, Kwan EK, Han SJ, Jin DK. Identification of mutations in the GNPTA (MFGC4170) gene coding for GlcNAc-phosphotransferase alpha/beta subunits in Korean patients with mucolipidosis type II and type IIIA. Hum Mut. 2005;26:308 - 14. [PubMed: 16116615]
- Raas-Rothschild A, Bargal R, Goldman O, Ben-Asher E, Groener JE, Toutain A, Stemmer E, Ben-Neriah Z, Flusser H, Beemer FA, Penttinen M, Olender T, Rein AJ, Bach G, Zeigler M. Genomic organization of the UDP-N-acetylglucosamine-1-phosphotransferase gamma subunit (GNPTAG) and its mutations in mucolipidosis III. J Med Genet. 2004;41:e52. [PMC free article: PMC1735719] [PubMed: 15060128]
- Raas-Rothschild A, Cormier-Daire V, Bao M, Genin E, Salomon R, Brewer K, Zeigler M, Mandel H, Toth S, Roe B, Munnich A, Canfield WM. Molecular basis of variant pseudo-hurler polydystrophy (mucolipidosis IIIC). J Clin Invest. 2000;105:673 - 81. [PMC free article: PMC289169] [PubMed: 10712439]
- Robinson C, Baker N, Noble J, King A, David G, Sillence D, Hofman P, Cundy T. The osteodystrophy of mucolipidosis type III and the effects of intravenous pamidronate treatment. J Inherit Metab Dis. 2002;25:681 - 93. [PubMed: 12705498]
- Spranger JW, Brill PW, Poznanski A. Mucolipidosis III. In: Bone Dysplasias: Atlas of Genetic Disorders of Skeletal Development. 2 ed. New York, NY: Oxford University Press; 2002:295-9.
- Steet RA, Hullin R, Kudo M, Martinelli M, Bosshard NU, Schaffner T, Kornfeld S, Steinmann B. A splicing mutation in the alpha/beta GlcNAc-1-phosphotransferase gene results in an adult onset form of mucolipidosis III associated with sensory neuropathy and cardiomyopathy. Am J Med Genet A. 2005;132:369 - 75. [PubMed: 15633164]
- Tappino B, Chuzhanova NA, Regis S, Dardis A, Corsolini F, Stroppiano M, Tonoli E, Beccari T, Rosano C, Mucha J, Blanco M, Szlago M, Di Rocco M, Cooper DN, Filocamo M. Molecular characterization of 22 novel UDP-N-acetylglucosamine-1-phosphate transferase alpha- and beta-subunit (GNPTAB) gene mutations causing mucolipidosis types IIalpha/beta and IIIalpha/beta in 46 patients. Hum Mutat. 2009;30:E956 - 73. [PubMed: 19634183]
- Tiede S, Muschol N, Reutter G, Cantz M, Ullrich K, Braulke T. Missense mutations in N-acetylglucosamine-1-phosphotransferase alpha/beta subunit gene in a patient with mucolipidosis III and a mild clinical phenotype. Am J Med Genet A. 2005;137A:235 - 40. [PubMed: 16094673]
- Zarghooni M, Dittakavi SS. Molecular analysis of cell lines from patients with mucolipidosis II and mucolipidosis III. Am J Med Genet A. 2009;149A:2753 - 61. [PubMed: 19938078]
Suggested Reading
- Ghosh P, Dahms NM, Kornfeld S. Mannose 6-phosphate receptors: new twists in the tale. Nat Rev Mol Cell Biol. 2003;4:202 - 12. [PubMed: 12612639]
- Gissen P, Maher ER. Cargos and genes: insights into vesicular transport from inherited human disease. J Med Genet. 2007;44:545 - 55. [PMC free article: PMC2597945] [PubMed: 17526798]
- Lachman R. Treatments for lysosomal storage disorders (2010). Biochem Soc Trans. 2010;38:1465 - 8. [PubMed: 21118108]
Chapter Notes
Revision History
- 10 May 2012 (me) Comprehensive update posted live
- 7 July 2009 (cd) Revision: deletion/duplication analysis available clinically for GNPTAB
- 26 August 2008 (cg) Review posted live
- 16 June 2008 (jgl) Original submission