
摘要
临床特征。
先天性基质角膜营养不良的特征是在出生时或出生后不久可见双侧角膜混浊。角膜表面正常或稍不规则;整个角膜基质中可见的小阴影使角膜呈浑浊状。斜视是常见的。眼球震颤并不常见。大多数人在青春期晚期或成年早期接受穿透性角膜移植术,效果良好。
诊断/检测。
DCN编码核心蛋白聚糖, 是唯一已知其致病性变异会导致先天性基质角膜营养不良的基因。
管理。
治疗表现:眼镜或隐形眼镜矫正屈光不正;修补和/或手术矫正斜视;穿透性角膜移植术。
遗传咨询。
先天性基质角膜营养不良以常染色体显性方式遗传。大多数被诊断患有先天性基质角膜营养不良的人有受累的父母。每个受影响的孩子有50%机会遗传致病性变异。如果已在受影响的家庭成员中鉴定出变异,通过实验室提供的产前基因检测包或定制化检测项目可以对高危妊娠进行产前检测。
诊断
临床诊断
先天性基质角膜营养不良的临床诊断是基于在出生时或出生后不久可见双侧角膜混浊(见图1):
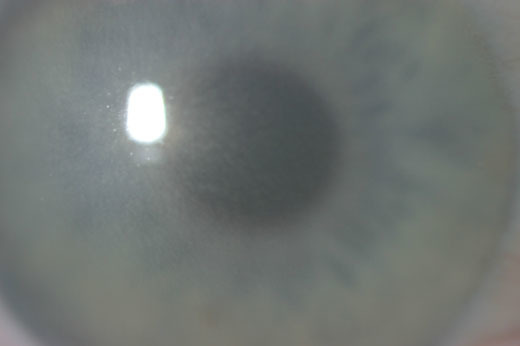 图1。
图1。角膜的裂隙灯照片显示角膜表面略不规则以及遍布整个角膜基质的小片状和小斑点。
- 角膜表面正常或稍不规则。
- 典型地,在整个角膜基质中可见的小阴影使角膜呈浑浊状。
- 角膜的厚度(用超声测厚法测量)通常是增加的。注意:这一发现可能有助于将先天的基质角膜营养不良与其他角膜厚度正常的疾病区分开。
- 眼压正常。
检测
基质的透射电子显微镜观察表明,正常胶原胶原纤维层被异常纤维层隔开,异常层中小单纤维嵌入了电子透明的基质中 (图2) [Bredrup et al 2005]。
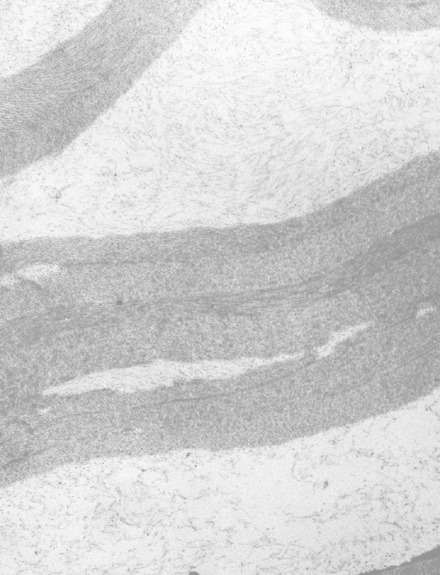
图2。
透射电子显微镜照片显示正常的胶原蛋白纤维片层,被具有薄纤维在电子透明基质中的异常层隔开。
分子遗传学检测
基因。DCN, 编码核心蛋白聚糖,是致病性变异导致先天性基质角膜营养不良的唯一基因 [Bredrup et al 2005, Rødahl et al 2006, Kim et al 2011].
Table 1.
先天性基质角膜营养不良的分子遗传学检测综述
| 基因 1 | 检测方法 | 致病性变异检测 | 通过这种方法检测到的致病变异频率3 |
|---|---|---|---|
| DCN | 序列分析 4 | 序列变异 | 100% 5 |
- 1.
请参阅表A基因和数据库的染色体位点和蛋白质。
- 2.
有关等位基因变异的信息,请参阅分子遗传学。
- 3.
检测方法用来检测特定基因的变异。
- 4.
序列分析可检测出良性,可能良性,意义不确定,可能致病性或致病性的的变异。致病性变异可能包括小的基因内缺失/插入和错义,无义和剪接位点变异;通常,未检测到外显子或全基因缺失/重复。有关在解释序列分析结果时要考虑的问题,请单击此处。
- 5.
序列分析迄今已经在三个家族中鉴定了DCN的致病性变异[Bredrup et al 2005, Rødahl et al 2006, Kim et al 2011]. 在三个家族中所有受累的 个体均具有一个 DCN 致病性变异。
临床特征
临床描述
文献中仅报道了具有先天性基质角膜营养不良(CSCD)的五个家族[Turpin et al 1939, Odland 1968, Witschel et al 1978, Van Ginderdeuren et al 2002, Kim et al 2011]. 在受影响的个体中已注意到一些家族间变异。
在一个有11个受累个体的挪威家庭中,在出生时或出生后不久观察到双侧角膜混浊[Bredrup et al 2005]。裂隙灯检查显示从角膜缘到角膜缘的的基质各层均有小片状、小斑点分布。角膜表面略微不规则。大多数患者的最佳矫正视力在0.63-0.3之间。11人中有4人患有斜视。没有人患有眼球震颤。角膜直径正常。测厚法显示角膜厚度增加(平均值:673μm;范围:658-704μm)。
受累个体的视力随着年龄的增长而下降;角膜混浊随年龄增加而增加。在平均年龄20岁的22只眼中有18只眼进行穿透性角膜移植。在3至36年的观察期内(平均19.5年),56%眼睛的移植片保持透明,另外33%的眼睛只有轻微的混浊。
在其他研究中,一些受累的个体被报道了畏光[Van Ginderdeuren et al 2002] 和眼球震颤 [Witschel et al 1978],后者最有可能是由于视力下降。正常角膜厚度通过测厚法也已有描述[Witschel et al 1978, Pouliquen et al 1979](尽管尚未得到证实)。
在其他器官系统中没有发现。
基因型-表型相关性
由于数据有限,没有明显的基因型-表型相关性。在 Lee et al [2012]报道的一个家族中,DCN致病性变异c.1036T>G与一种类似CSCD的相对温和的迟发性疾病有关。
外显率
在所描述的家庭中,外显率是完全的。
术语
先天性基质角膜营养不良的其他名称:
- 先天性实质性角膜营养不良
- 先天性角膜基质营养不良
发病率
CSCD可能非常罕见。 已经描述了五个具有相似表型的家族。其中的三个,分子分析揭示了DCN致病变异(表2)[Bredrup et al 2005, Rødahl et al 2006, Kim et al 2011]。
遗传相关(等位基因)疾病
尚无其他与DCN致病变异相关的表型。
鉴别诊断
双侧先天性角膜混浊可由多种疾病引起,包括:
- 先天性青光眼
- 眼前节畸形
- 炎症
- 全身性贮积病
- 各种角膜营养不良[ Weiss et al 2015 ],主要是先天性遗传性内皮营养不良(OMIM 217700)
疾病管理
初步诊断后的评估
建立个体诊断出患有先天性基质角膜营养不良的疾病程度,眼科评估包括以下建议:
- 视力评估
- 屈光不正的评估
- 裂隙灯检查
- 用测厚仪测量角膜厚度
- 眼压测量
- 临床遗传学咨询
治疗表现
以下是适当的:
- 眼镜或隐形眼镜用于矫正屈光不正
- 斜视管理
- 穿透性角膜移植术。大部分的移植角膜在穿透性角膜移植术后保持清晰。10岁以下儿童穿透性角膜移植已取得成功。然而,只有有深度弱视风险的儿童才应该考虑在六、七岁之前进行穿透性角膜移植。
继发性并发症的预防
对斜视儿童进行修补以预防弱视。
监护
儿童应至少每年进行视力检查和常规眼科检查。除非进行了穿透性角膜移植,否则无需对成人进行定期监护。受影响的个体应被告知穿透性角膜移植术,并建议在视力下降或眩光增加时联系他们的眼科医生。
家属患病风险的评估
在患有CSCD的家庭中,眼科医师应在分娩后的几个月内对高危儿童进行检查,以确定他们是否患有这种疾病。或者,如果已确定该家庭中的DCN 致病性变异,则可以进行高危儿童的分子遗传学检测。
患者亲属患病风险的遗传咨询以及相关检测信息参见 遗传咨询 。
调查研究中的治疗方法
搜索ClinicalTrials.gov ,以获取有关各种疾病和状况的临床研究信息。注意:可能没有该疾病的临床试验。
遗传咨询
遗传咨询是为个人和家庭提供有关遗传性疾病的本质,遗传特性和影响的信息,以帮助他们做出知情的医疗和个人决定的过程。以下部分讨论遗传风险评估以及根据家族史和遗传检测来阐明家族成员的遗传状况。本部分不适用于解决个人可能面临的所有个人、文化或伦理问题,也不能代替专业的遗传咨询。—ED.
遗传模式
先天性基质角膜营养不良(CSCD)以常染色体显性方式遗传。
家族成员患病风险
先证者的父母
- 大多数被诊断为先天性基质角膜营养不良的个体都有受累的父母。
- 先天性基质角膜营养不良的先证者可能由于新的致病性变异而患有疾病。新生致病性变异引起的病例比例未知。
- 父母的评估可能是其中一个是受累的,但因为温和的表型之前未被诊断。因此,在进行适当的临床评估之前,无法确认明确的阴性家族史。
先证者的同胞
- 先证者同胞的风险取决于先证者父母的遗传状况。
- 如果先证者的父母是受累的,同胞的遗传风险是50%概率。
- 当父母临床上未受影响,先证者同胞的患病风险低。
- 虽然未被报道,但先证者父母外显率减少或胚系嵌合理论上是可能的;因此,临床上未受影响的父母的先证同胞的患病风险可能仍然会增加。
先证者的子代。 患有先天性基质角膜营养不良个体的孩子有50%的概率会遗传DCN致病变异。
先证者的其他家族成员。对其他家庭成员的风险取决于先证者父母的状况:如果父母是受累的,则他或她的家庭成员可能会面临风险。
相关的遗传咨询问题
有关为早期诊断和治疗目的评估高危亲属的信息,请参阅管理,家属患病风险的评估(Evaluation of Relatives at Risk)。
有明显的家庭新生致病性变异(de novo 致病性变异)的考虑。当具有常染色体显性遗传疾病的先证者的父母均没有该疾病的临床证据时,先证者很可能是新生致病性变异。然而,也可以探索可能的非医学解释包括非生物学父亲或母亲(例如辅助生殖)或秘密收养。
家庭计划
- 确定遗传风险和讨论产前检测可用性的最佳时间是在怀孕之前。
- 向受累或有风险的年轻人提供 遗传咨询(包括对对后代和生殖选择潜在风险的讨论)是适当的。
DNA库是DNA(通常从白细胞中提取)的存储,以备将来使用。由于检测技术以及我们对基因、等位基因变异和疾病的理解在未来将深入,因此受累的个体可考虑基因库。
产前诊断和胚胎植入前遗传学检测
一旦在受累的家庭成员中发现了DCN 致病性变异,就可以对高危妊娠进行产前检测和选择植入前遗传诊断。
要求产前检测(如先天性基质角膜营养不良)不影响才智和有一些治疗可得的情况并不常见。在使用产前检测方面,医学专业人员和家庭内部可能存在着不同的看法,特别是如果考虑将产前检测用于终止妊娠而不是早期诊断的情况下。尽管大多数中心将有关产前检查的决定视为父母的选择,但对这些问题的讨论是适当的。
资源
GeneReviews工作人员已经筛选了以下专科疾病和/或orumbrella帮扶组织和/或登记处,以使这种疾病的患者及其家人受益。GeneReviews对该类组织提供的信息不负责,信息的筛选标准,参见此处here。
- National Eye Institute31 Center DriveMSC 2510Bethesda MD 20892-2510Phone: 301-496-5248Email: 2020@nei.nih.gov
- National Eye Institute31 Center DriveMSC 2510Bethesda MD 20892-2510Phone: 301-496-5248Email: 2020@nei.nih.gov
分子遗传学
分子遗传和OMIM表中的信息可能与GeneReview中的其他信息不同:表格可能包含更多最新信息 - ED.
Tabl表A.e A.
先天性基质角膜营养不良:基因和数据库
Table B.
先天性基质角膜营养不良的OMIM条目 ( 在OMIM中查看全部)
分子遗传学发病机理
角膜透明度要求胶原纤维被适当地组织成具有均匀的直径和规则的原纤维间空间。先天性间质角膜营养不良(CSCD)的特征是整个角膜基质混浊。通过透射电子显微镜,这些混浊被视为具有细肌丝的无定形材料。已报道的DCN致病变异均导致一种截短的核心蛋白聚糖的形成,这种蛋白在体外具有聚集的趋势。核心蛋白聚糖会在无定形区聚集。作者假设截短的核心蛋白聚糖会在CSCD中积聚,从而导致混浊[Bredrup et al 2010]。
基因结构。DCN 基因长度 3777 kb. 全长基因由8个外显子组成,外显子2中带有AUG起始密码子。有关基因和蛋白质信息的详细摘要,请参见 表A, 基因。
良性等位基因变异。1号内含子中存在不完美的二核苷酸重复变异。在一小群1型糖尿病患者中,这些变异之一与肾脏疾病的缓慢进展有关[De Cosmo et al 2002](见表A,HGMD)。
致病性等位基因变异。在DCN中已检测到3个变异与先天性基质角膜营养相关(表2) [Bredrup et al 2005, Rødahl et al 2006, Kim et al 2011]。全部移码变异位于最后一个编码外显子。所得的蛋白质预测具有几个末端氨基酸残基的改变和33个C末端氨基酸的缺失。
Table 2.
本章所选的DCN 致病性等位基因变异
| DNA核苷酸变化 | 预测的蛋白质变化 | 参考序列 |
|---|---|---|
| c.967delT | p.Ser323LeufsTer5 | NM_001920 NP_001911 |
| c.941delC | p.Pro314HisfsTer14 | |
| c.947delG | p.Gly316AspfsTer12 |
关于变异分类的注意事项:表中列出的变异由作者提供。GeneReviews工作人员尚未独立验证变异的分类。
关于命名的注意事项:GeneReviews是依照人类基因组变异协会 (varnomen
- .hgvs.org )的标准命名规范。有关命名法的说明,请参见Quick Reference。
正常基因产物。核心蛋白聚糖(Decorin)是富含亮氨酸的小重复蛋白聚糖的I类家族成员;此类的其他成员包括双糖链蛋白聚糖和无孢蛋白。 从N端切割出一个14个氨基酸的前肽,形成329个氨基酸的最终“成熟”核心蛋白。“成熟”的核心蛋白在p.Ser4残基处以及p.Asn181,p.Asn232和p.Asn273残基的两个或三个N-连接的寡糖处被一个硫酸软骨素/硫酸皮肤素糖胺聚糖链取代。核心蛋白聚糖(Decorin)广泛分布于结缔组织中,可与几种生物学上重要的分子结合,包括胶原蛋白I,胶原蛋白VI,纤连蛋白,血小板反应蛋白,表皮生长因子受体,胰岛素样生长因子1受体和转化生长β因子。它参与了许多生物学过程,主要是胶原纤维形态的调节,也参与了细胞粘附、血管生成、细胞基质形成和细胞增殖的调控[Schaefer & Iozzo 2008]。有证据表明,核心蛋白聚糖是胶原纤维横向生长的主要抑制剂[Zhang et al 2009]。除了作为一种结构蛋白的既定作用外,核心蛋白聚糖还可能作为一种调节蛋白发挥重要作用。
异常基因产物。 目前在DCN中检测到的3种致病性移码变异会导致一些氨基酸的改变和蛋白质的过早截短(如33个羧基末端氨基酸的截短)[Bredrup et al 2005, Rødahl et al 2006, Kim et al 2011]。预测的截断蛋白被假设为会导致核心蛋白聚糖在角膜中积累,从而导致角膜混浊。其积累机制可能是核心蛋白聚糖的聚集。
注意:核心蛋白聚糖(Decorin)在恶性肿瘤中引起了特别的关注,因为核心蛋白聚糖已被证明是一种强的细胞生长抑制剂,并起促凋亡作用。这些研究都没有涉及人类的胚系变异。
参考文献
文献引用
- Bredrup C, Knappskog PM, Majewski J, Rødahl E, Boman H. Congenital stromal dystrophy of the cornea caused by a mutation in the decorin gene. Invest Ophthalmol Vis Sci. 2005;46:420 - 6. [PubMed: 15671264]
- Bredrup C, Stang E, Bruland O, Palka BP, Young RD, Haavik J, Knappskog PM, Rødahl E. Decorin accumulation contributes to the stromal opacities found in congenital stromal corneal dystrophy. Invest Ophthalmol Vis Sci. 2010;51:5578 - 82. [PubMed: 20484579]
- De Cosmo S, Tassi V, Thomas S, Piras GP, Trevisan R, Cavallo Perin P, Bacci S, Zucaro L, Cisternino C, Trischitta V, Viberti GC. The decorin gene 179 allelic variant is associated with a slower progression of renal disease in patients with type 1 diabetes. Nephron. 2002;92:72 - 6. [PubMed: 12187087]
- Kim JH, Ko JM, Lee I, Kim JY, Kim MJ, Tchah H. A novel mutation of the decorin gene identified in a Korean family with congenital hereditary stromal dystrophy. Cornea. 2011;30:1473 - 7. [PubMed: 21993463]
- Krachmer JH, Mannis MJ, Holland EJ, eds. Cornea. Vol 1. 2 ed. Philadelphia, PA: Elsevier Mosby; 2004.
- Odland M. Dystrophia corneae parenchymatosa congenita. A clinical, morphological and histochemical examination. Acta Ophthalmol (Copenh) 1968;46:477 - 85. [PubMed: 5304426]
- Pouliquen Y, Lacombe E, Schreinzer C, Giraud JP, Savoldelli M. Familial congenital dystrophy of the corneal stroma: Turpin's syndrome (author's transl) J Fr Ophtalmol. 1979;2:115 - 25. [PubMed: 312637]
- Rødahl E, Van Ginderdeuren R, Knappskog PM, Bredrup C, Boman H. A second decorin frame shift mutation in a family with congenital stromal corneal dystrophy. Am J Ophthalmol. 2006;142:520 - 1. [PubMed: 16935612]
- Schaefer L, Iozzo RV. Biological functions of the small leucine-rich proteoglycans: from genetics to signal transduction. J Biol Chem. 2008;283:21305 - 9. [PMC free article: PMC2490788] [PubMed: 18463092]
- Turpin R, Tisserand M, Sérane J. Opacités cornéennes héréditaires et congénitales réparties sur trios générations et atteignant deux jumelles monozygotes. Arch Ophthalmol (Paris) 1939;3:109 - 11.
- Van Ginderdeuren R, De Vos R, Casteels I, Foets B. Report of a new family with dominant congenital heredity stromal dystrophy of the cornea. Cornea. 2002;21:118 - 20. [PubMed: 11805522]
- Witschel H, Fine BS, Grützner P, McTigue JW. Congenital hereditary stromal dystrophy of the cornea. Arch Ophthalmol. 1978;96:1043 - 51. [PubMed: 350201]
- Zhang G, Chen S, Goldoni S, Calder BW, Simpson HC, Owens RT, McQuillan DJ, Young MF, Iozzo RV, Birk DE. Genetic evidence for the coordinated regulation of collagen fibrillogenesis in the cornea by decorin and biglycan. J Biol Chem. 2009;284:8888 - 97. [PMC free article: PMC2659246] [PubMed: 19136671]
推荐阅读
- Ciralsky J, Colby K. Congenital corneal opacities: a review with a focus on genetics. Semin Ophthalmol. 2007;22:241 - 6. [PubMed: 18097987]
- Weiss JS, Møller HU, Lisch W, Kinoshita S, Aldave AJ, Belin MW, Kivelä T, Busin M, Munier FL, Seitz B, Sutphin J, Bredrup C, Mannis MJ, Rapuano CJ, Van Rij G, Kim EK, Klintworth GK. The IC3D classification of corneal dystrophies. Cornea. 2008;27 Suppl 2:S1 - 83. [PMC free article: PMC2866169] [PubMed: 19337156]
章节注释
修订记录
- 2 February 2012 (me) Comprehensive update posted live
- 25 November 2008 (me) Review posted live
- 10 September 2008 (er) Original submission