
概要
临床特征.
最典型的是Aicardi-Goutières综合症(AGS)表现为一种早发性脑病,通常但并非总是导致严重的智力和身体残疾。一组AGS亚组的婴儿出生时具有异常的神经系统检查结果,肝脾肿大,肝酶升高和血小板减少症, 很像 先天的感染。其他大部分受累的 婴儿经常在生命的最初几周、表面上发育正常之后才出现症状。通常,他们表现出严重脑病的亚急性发作,其特征为极度易怒,间歇性无菌性发热,技能丧失和头部生长减慢。随着时间的流逝,多达40%的人在手指,脚趾和耳朵上出现冻疮样皮肤损伤。显而易见的是,存在非典型的,有时较温和的AGS病例,因此,与AGS相关基因中的致病变异相关的表型的真实程度尚不清楚。
诊断/测试.
AGS的诊断是建立在有以下症状的先证者基础之上 :
- 具有典型的临床发现以及有异常特征性的颅脑CT(基底神经节和脑白质钙化)和MRI(白质营养不良性改变)的;
- 并有或者有以下之一来特点来诊断:
- 在 ADAR, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, or TREX1存在双等位基因致病变异
管理.
duiz治疗:胸部理疗和呼吸系统并发症的治疗;注意饮食和喂养方法,以确保摄入足够的热量并避免误吸;使用标准方案管理癫痫发作。
监测:监测新生儿期尿崩症的体征;至少在生命的最初几年重复眼科检查,以评估青光眼的证据;监测脊柱侧弯,胰岛素依赖型糖尿病和甲状腺功能减退的证据。
遗传咨询.
AGS最常以常染色体隐性遗传 。在少数情况下,该疾病可能是由于ADAR或TREX1中特定的新发生/de novo 或遗传性常染色体显性遗传致病性变异,以及IFIH1中多种杂合的 常染色体显性致病性变异引起的。在受孕时,患有常染色体隐性遗传的AGS受累的个体的每个同胞都有25%的机会受到感染,有50%的机会成为无症状携带者,并且有25%的几率不受影响也不是携带者。具有AGS的个体通常不会生育。一旦在受影响的家庭成员中确定了致病变异,就可以选择对高危亲属进行 携带者检测,对AGS风险增加的妊娠进行产前检测,以及进行植入前基因检测/preimplantation genetic testing.
诊断
最典型的Aicardi-Goutières综合征(AGS),可被视为与严重智力和身体残疾相关的早发性脑病。
提示性发现
有以下临床表现,神经影像学和支持性实验室检查结果的个体应怀疑患有Aicardi-Goutières 综合征(AGS) [Goutières et al 1998, Lanzi et al 2002, Rice et al 2007b, Livingston et al 2013].
临床特征
- 脑病和/或严重智力障碍
- 出生后第一年获得小头畸形
- 肌张力障碍和痉挛
- 无菌性发热
- 肝脾肿大
- 脚,手,耳朵的冻疮样病变,更多的时候为一般皮肤斑点。见 Figure 1.
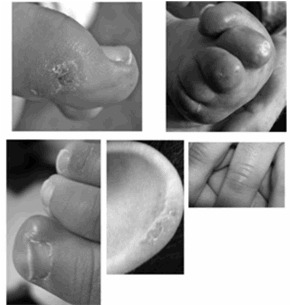
Figure 1.
AGS Rice等人[2007b]图中所见的冻疮样病变的例子。转载经《美国人类遗传学杂志》,芝加哥大学出版社。
排除标准包括以下内容:
- 产前/围产期感染的证据,包括但不限于CMV,弓形虫病,风疹,单纯疱疹,寨卡病毒和HIV
- 已知的其他代谢性疾病或神经退行性疾病的证据
神经影像学
- 基底节的钙化(最好在CT扫描上可见),尤其是壳核,苍白球和丘脑的钙化,但也延伸到白质中,有时呈旁心室分布(而不是真正分布在周围) [Lanzi et al 2002, Uggetti et al 2009, Livingston et al 2013]. See Figure 2。
注意:颅内钙化并不总是能在MRI上识别,MRI是大多数单位使用的初始成像方式。
在MRI上,T 2加权图像上出现的高强度信号通常位于脑室角周围(图3BFigure 3B)
- 脑萎缩,可能是进行性的,累及脑室周围白质和脑沟
小脑萎缩和脑干萎缩也可能很明显(图3D/Figure 3D) [Crow et al 2004a, Sanchis et al 2005]。
- 双侧纹状体坏死
- 脑内血管病变,包括颅内狭窄,烟雾病和动脉瘤
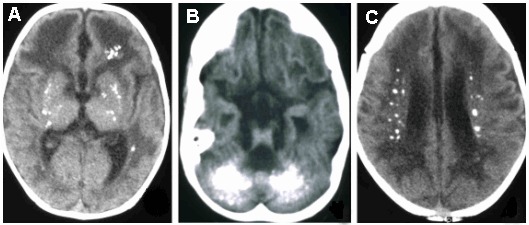
Figure 2.
AGS患者CT扫描中颅内钙化的例子。可见钙化:A.在基底神经节中;
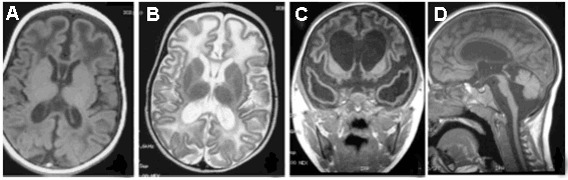
Figure 3.
在AGS A的MRI中看到的脑部变化的频谱。白质T 1加权成像的低敏性
支持性实验室检查
外周血
- 使用RNA / cDNA的quantitative PCR分析鉴定出干扰素阳性[ Rice et al 2013a, Crow et al 2015 ]
- 肝酶升高
- 血小板减少症
脑脊液 (CSF)
- 慢性CSF白细胞增多症,定义为超过5个淋巴细胞/ mm 3 CSF。
- 典型值范围是从5到100个淋巴细胞/ mm 3 [ Goutières et al 1998, Rice et al 2007b, Rice et al 2013a]。
- 淋巴细胞数量会随着时间而减少,尽管高细胞数可能会持续数年。
- 即使在疾病早期,在脑脊液中存在升高浓度的IFN-α时,也可以观察到正常细胞计数[ Crow et al 2003, Rice et al 2007b ]。
- 脑脊液中干扰素-α(IFN-α)活性增加(正常值:<2 IU / mL)
- 在疾病的早期阶段,记录的IFN-α活性通常最高。IFN-αCSF活性可以在生命的最初三到四年内恢复正常[ Rice et al 2007b, Rice et al 2013a]
- 在脑脊液中记录的IFN-α活性通常高于血液,后者可能是正常的。
- 妊娠26周时在胎儿血液中发现了高IFN-α活性[ Desanges et al 2006 ]。
- 脑脊液中新蝶呤浓度的增加[ [Rice et al 2007b]
- 该水平在疾病的早期阶段最高,并且可以随着时间恢复正常。
- 神经递质代谢产物5HIAA,HVA和5MTHF的水平正常。
建立诊断
AGS的诊断是建立在一个先证者典型的临床表现和特性的异常颅CT(基底神经节钙化和白质)和MRI(白质营养不良改变)和/或通过的发现ADAR,RNASEH2A,RNASEH2B,RNASEH2C,SAMHD1或TREX1基因上的双等位基因的致病变异 ; 在TREX1和ADAR中有特定的常染色体显性杂合子致病变异; 或IFIH1中的各种杂合的 常染色体显性遗传的致病变异。
分子检测方法可以包括一系列单基因检测,多基因套餐/ multigene panel检测,以及使用更全面的基因组的 检测。
可以根据个人的种族和/或按照最常见的病原体变异顺序进行系列单基因检测(请参见表1/ Table 1)。
- 如果只有一个 致病性变异在确定RNASEH2A,RNASEH2B,RNASEH2C,或SAMHD1,基因靶向性缺失/重复分析/deletion/duplication analysis 可接下来考虑。
- 如果在TREX1或ADAR中发现了未知的与 常染色体显性遗传AGS相关的杂合的 致病性变异,也可以考虑针对基因的缺失/重复分析/deletion/duplication analysis。
一个多基因套餐检测/multigene panel ,其包括ADAR,IFIH1,RNASEH2A,RNASEH2B,RNASEH2C,SAMHD1,TREX1,和感兴趣的其它基因(见Differential Diagnosis/鉴别诊断)也可以被考虑。注意:(1)所包含的基因和多基因组的敏感性因实验室而异,并可能随时间变化。(2)一些多基因组可能包含与本GeneReview中讨论的病症无关的基因; 因此,临床医生需要确定哪个多基因小组最有可能以最合理的成本鉴定出该病的遗传原因,同时限制了对意义不确定的变异和无法解释潜在表型的基因中的致病性变体的鉴定。(3)在某些实验室中,套餐选项可能包括定制的实验室设计套餐和/或定制的以表型为中心的外显子组 分析,其中包括临床医生指定的基因。(4)专家组中使用的方法可能包括序列分析, deletion/duplication analysis/删除/重复分析和/或其他基于非序列的测试。
有关多基因套餐的介绍,请单击此处/ here。有关订购基因检测的临床医生的更多详细信息,请参见此处/here。
更全面的基因组的 测试(当可用时),包括外显子组测序,线粒体测序和基因组测序可以认为如果一系列单基因测试(和/或使用的multigene panel/多基因套餐,其包括ADAR,IFIH1,RNASEH2A,RNASEH2B,RNASEH2C,SAMHD1,和TREX1)无法确认具有AGS特征的个体的诊断。这样的测试可以提供或提示先前未考虑的诊断(例如,导致相似临床表现的一个或多个不同基因的突变)。有关全面的基因组测试的介绍,请单击here。可在here./此处找到有关订购基因组测试的临床医生的更多详细信息。
表格1。Aicardi-Goutières 综合征的分子遗传学检测 Table 1.
基因 1 | 归因于基因致病变异的AGS的比例 | 方法可检测的致病变异体2比例 | |
序列分析 3,4 | |||
ADAR | 7% | 32/32等位基因位点 6 | 未知 7 |
IFIH1 | 3% | 17/17等位基因位点 8 | 未知 7 |
RNASEH2A | 5% | 34/34等位基因位点 9 | 未知 7 |
RNASEH2B | 36% | 〜99% 10 | 未知 11 |
RNASEH2C | 12% | 〜99% 12,13 | 未知 7 |
SAMHD1 | 13% | 高达95% 14 | 高达30% 14,15 |
TREX1 | 23% | 〜99% 16 | 未知 7 |
未知 | 1% | NA | |
1。请参阅表A基因和数据库/Table A. Genes and Databases的染色体 基因和蛋白质。
2。有关在该基因中检测到的等位基因变体的信息,请参见《分子遗传学/Molecular Genetics》。
3。序列分析可检测出意义不佳,可能是致病性或致病性的良性,可能良性的变体。致病性变异可能包括小的基因内缺失/插入和 错义, nonsense, 和 剪接位点变异;通常,未检测到外显子或全基因缺失/重复。有关在解释序列分析结果时要考虑的问题,请单击此处 here。
4。经过ADAR,IFIH1,RNASEH2A,RNASEH2B,RNASEH2C,SAMHD1和TREX的编码区和剪接位点的序列分析,在约90%-95% 有临床表现和MRI特征的AGS病例中,已确定了致病变异-Rice et al 2007b, Rice et al 2009, Crow et al 2015 ]。
5,基因靶向的缺失/重复分析 deletion/duplication analysis检测基因内的缺失或重复。所使用的方法可能包括 quantitative PCR/定量PCR,长距离PCR,多重连接依赖性探针扩增(MLPA),以及旨在检测单外显子缺失或重复的基因靶向微阵列。
6。通过序列分析可检测到32/32个等位基因 [Crow et al 2015]。已经报道了ADAR中主要的 p.Gly1007Arg变异[Rice et al 2012, Livingston et al 2014a ]。在源自欧洲的受累的人群中发现了一种复发性ADAR变异 p.Pro193Ala。
7。没有涉及ADAR,IFIH1,RNASEH2A,RNASEH2C,或TREX1的缺失或重复被报道引起Aicardi-Goutières综合征。
8。Rice et al [2014], Crow et al [2015]
9。Rice et al [2007b], Crow et al [2015]
10。在RNASEH2B中具有 双等位基因的致病变异的个体中,有90%为错义变化的纯合性 或 复合杂合 子(p.Ala177Thr)。
11。缺失的RNASEH2B两个通5个外显子有报道[[Crow et al 2015]。
12. Rice et al [2007b], Crow et al [2015]
13.该RNASEH2C变异p.Arg69Trp,尤其常见于亚洲(最常见的巴基斯坦)的家庭,并代表一种古老的建立者变异[Rice et al 2007b ]。
14。通过序列分析检测到25/26个等位基因[Rice et al 2009 ]和47/67个等位基因[ Crow et al 2015 ] 。注意:Rice et al [2009] 未进行缺失/重复分析deletion/duplication analysis ,在本研究的某些家族中未检测到致病变异。
15.在 Ashkenazi 犹太血统的几个受累的个体中观察到包括外显子1在内的反复发生的缺失,很可能代表了一个建立者变异[ Ramesh et al 2010 ]。可通过缺失/重复分析 deletion/duplication analysis检测到20/67个中的等位基因 Crow et al [2015] ;然而,这项研究包括了阿什肯纳兹犹太血统的人。注意:利用PCR扩增不能检测到的外显子,可以怀疑≥1个外显子的纯合性纯合缺失。在阿米什人的祖先中可以看到12号内含子 中一个复发的剪接受体位点致病性变异 (c.1411-2A>G),代表了一个古老的建立者变异[Xin et al 2011]。
16。在大约100个与TREX1相关的AGS的个体中,通过序列分析 [Rice et al 2007b, Crow et al 2015]可检测到所有致病变异]。在TREX反复出现的p.Arg164Ter 建立者变异1被认为是来自克里的祖先个体。AGS中最流行的TREX1变异是一种错义变化(p.Arg114His) ,在北欧人中尤为常见。
- 16.
临床特征
临床描述
最典型的Aicardi-Goutières综合征(AGS)可被视为与严重智力和身体残疾相关的早发性脑病。
孕妇的妊娠,分娩和新生儿期 约80%的患有Aicardi-Goutières综合征(AGS)的婴儿表现是正常的[ Rice等,2007b ]。然而,也可以在子宫内发现脑钙化[ Le Garrec et al 2005 ],主要是由TREX1中的双等位基因致病变异引起的,在出生时出现异常的神经系统发现,肝脾肿大,肝酶升高和血小板减少,临床上与先天性感染很像。
所有其他 受累的婴儿在生命的最初几周好像发育正常,以后陆续才出现症状。这些后发病的婴儿大多数表现出严重急性脑病的亚急性发作,其特征是极度易怒,间歇性无菌性发热,技能丧失和头部生长减慢。脑病阶段通常持续数月。大多数照料此类儿童的儿科医生的意见是,这种疾病不会在脑病期后再加重。但是,有时患儿似乎确实显示出进展和/或消退发作。通常认为死亡是继发于初始疾病发作期间的神经系统损害的继发原因,而不是进一步退行性病变。
神经学特征。通常, 受累的个体具有周围性痉挛,肌张力低下的姿势(尤其是上肢),躯干性肌张力低下和头部控制不佳。据报道,多达一半的患儿有癫痫发作,但通常相对容易控制[ Goutières等,1998;Rice等,2007b ]。许多孩子表现出对突然发出的噪音有明显的惊吓反应,并且很难与癫痫病鉴别。大多数患者有严重的智力和身体障碍。同胞之间可观察到神经系统预后的严重程度差异。大多数受影响的儿童表现出严重的后天性小头畸形。保留智商的儿童的头围可以正常。
听觉几乎总是正常的。
眼科。视觉功能从正常到皮层失明。眼部检查几乎总是正常的。然而,存在先天性青光眼或迟发性青光眼的风险[ Crow et al 2004a,Crow et al 2015 ]。
皮损。多达40%的受感染个体[ Rice等,2007b ]出现皮肤病变,手指和脚趾上有时有耳朵上有冰斑,耳朵和其他压力点(例如肘部)[ Tolmie等,1995;Stephenson,2002 ](见图1)。皮肤病变可能并发唇周感染和坏死。
其他
- 颅内大血管疾病。迄今为止,AGS的另一个以前未描述的特征似乎几乎完全与SAMHD1中的双等位基因致病变异有关,是引起颅内狭窄(在某些情况下让人联想到Moyamoya综合征)和动脉瘤的颅内大血管疾病(请参阅基因相关性表型/ Phenotype Correlations by Gene[ Ramesh等人2010,Thiele等人2010,du Moulin等人2011,Xin等人2011 ]。
- 难治性四肢肌张力障碍。据报道,一些具有ADAR致病性变异的个体表现出从8个月至5岁开始的难治性四肢肌张力障碍的急性或亚急性发作[ Livingston等,2014a,Crow等,2015 ](见基因相关性表型)
表2总结了AGS的其他不常见特征。 Table 2/表2 在队列确认的123个人中发现的罕见特征
特征 | 受基因影响的个体数量 | |||
RNASEH2A | RNASEH2B | RNASEH2C | TREX1 | |
脊柱侧弯 | 0 | 9 | 0 | 0 |
心脏肥大 | 1个 | 0 | 1个 | 4 |
抗体异常 | 0 | 3 | 1个 | 2 |
保留语言 | 0 | 6 | 0 | 0 |
脱髓鞘性周围神经病 | 0 | 2 | 1个 | 1个 |
先天性青光眼 | 0 | 0 | 1个 | 2 |
小阴茎 | 0 | 0 | 1个 | 1个 |
甲状腺功能减退 | 0 | 1个 | 0 | 1个 |
胰岛素依赖型糖尿病 | 0 | 1个 | 0 | 1个 |
抗利尿激素的短暂性缺乏 | 0 | 0 | 0 | 1个 |
Rice et al [2007b].注意:在确定ADAR,IFIH1和SAMHD1中的致病变异可导致AGS之前,已发表了这篇论文。
神经影像/神经病理学发现
- 神经影像学的钙化:
- 常呈点状,但可能密集、像石头;
- 尽管可以观察到进展,但在诊断时被鉴定后往往会保持稳定[ Lanzi等,2002;Lanzi等,2005 ]。
- 与神经系统预后的严重程度无关;在极少数情况下,可能不存在钙化,并且在重复扫描中可能看不到钙化。因此,没有钙化并不排除诊断[ Aicardi&Goutières1984 ]。
- 在受严重受累的个体中发现的主要神经病理学发现包括[ Kumar等1998,Barth 2002 ]:
- 弥漫性但非均质性脱髓鞘伴星形胶质细胞增多症;没有储存迹象或髓磷脂降解;
- 新皮层和小脑皮层多发楔形微梗塞,提示微血管病变。
- 白质,丘脑,基底神经节和齿状核中有钙质沉积;
- 小血管的中膜,外膜和血管周围空间的钙化;
- 轻发和坏死部位发炎。
基因相关的表型
在一般情况下,AGS的早期发作新生儿形式多见于几种 双等位基因的致病变异RNASEH2A,RNASEH2C,或TREX1。较晚出现的表现(有时在正常发育的几个月后发生,偶尔与神经功能显着相关)在RNASEH2B,SAMHD1或ADAR中与双等位基因致病变体有关,但也可能见于常染色体显性遗传杂致病变种在ADAR或IFIH1 [ Crow et al 2015 ]。
死亡率与基因型相关:有RNASEH2A,RNASEH2C和TREX1致病变异的患者,其死亡率34%,相比 RNASEH2B致病变异的患者死亡率仅8%(p值= 0.001)[ Rice等人,2007年b ]。与其他基因型相关的死亡率尚不清楚。
ADAR。ADAR的致病变异与急性双侧纹状体坏死的临床表现有关。
具有ADAR致病性变体的一个亚组的个体可表现为尾状核和壳核的对称性信号改变,通常与急性或亚急性难治性四肢肌张力障碍的发生有关。这些病例在发育完全正常的背景下,最晚可在4岁时发病具有ADAR致病性变体的亚组个体可能在尾状核和壳状核中出现对称的信号变化,在难治性四肢肌张力障碍的急性或亚急性发作情况下,通常与肿胀和继发的收缩有关。在完全正常发展的背景下,已鉴定出发病至四岁的病例[ La Piana等,2014;Livingston等,2014a ]。
鉴别诊断为非症状性双侧纹状体坏死(BSN)伴有不同演变的严重肌张力障碍时,应考虑与ADAR相关的疾病。在这种情况下,干扰素标记的发现为ADAR致病性变体的存在提供了有用的筛选测试。
RNASEH2B。一些RNASEH2B具有双等位基因致病变异的患者相对保留着正常的智力功能,少数个体具有完全正常的智商和头围[ McEntagart等1998 ]。
RNASEH2C。已经描述了一个家庭[Crow et al 2015]携带有亚洲反复出现的RNASEH2C中的奠基者变异,且发病情况在该家系内具有差异。
SAMHD1
- 脑内血管病变,包括颅内狭窄和动脉瘤,在具有SAMHD1中双等位基因致病变异的个体中更为常见。
- Dale等人[2010]报道了两个在SAMHD1中具有双等位基因 无效变异的同胞。年龄较大的女孩表现出轻度智力障碍,伴小头畸形。她的弟弟患有明显的痉挛性轻瘫,智力和头部大小正常。两个孩子都患有未分类的慢性炎症性皮肤病,伴有冻疮样皮损和口腔溃疡反复发作。其中一个同胞患有继发性挛缩的小关节和大关节的慢性进行性变形关节炎。Ramantani等人[2010]也描述了类似的关节受累(另见Crow等人[2015])。
- SAMHD1中的双等位基因致病变异最常与冻疮和青光眼相关[ Crow et al 2015 ]。
命名法
小头颅内钙化综合征(MICS;也称为伪TORCH综合征或Baraitser-Reardon综合征)以前是根据先天性小头畸形和非神经系统异常(包括肝酶升高、肝脏肿大和出生时的血小板减少症与AGS区分的[ Reardon等1994 ]。然而,最近的研究表明,在已经鉴定出相关基因之一的致病变异的AGS患者中也可以看到这些相同的特征[ Rice等,2007b ]。值得注意的是,在大多数MICS病例中,尚无关于CSF细胞计数和IFN-α浓度的信息。因此,大多数MICS案例实际上都是AGS。
“家族性系统性红斑狼疮。” Dale等人[2000]描述了两个近亲结婚父母的孩子,他们患有早发性脑病,颅内钙化,冻疮皮肤病变,并逐渐产生高水平的自身抗体。没有分析脑脊液。这些案例最有可能代表AGS [ Aicardi&Goutières2000 ]。还描述了具有类似系统性红斑狼疮的症状和体征的其他具有AGS的个体。
患病率
AGS的实际频率未知。
在所有种族血统的 受累的个体中都发现了致病变异[ Crow等人2006a,Crow等人2006b,Rice等人2007b,Rice等人2013b ](参见表1)。
遗传相关疾病(等位基因)
表3列出了由ADAR,IFIH1,RNASEH2B和TREX1中的病原体变异引起的其他表型。
等位基因疾病
疾病名 | ADAR | IFIH1 | RNASEH2B | SAMHD1 | TREX1 1 |
对称性色素沉着病1(DSH) 2 | X | ||||
“纯”痉挛性轻瘫 3或儿童期发作性轻瘫4智力正常 | X | X | X | ||
常染色体显性遗传性视网膜血管病伴脑白质营养不良(RVCL) 5 | X | ||||
系统性红斑狼疮(SLE) 6 | X | X | X | ||
克里(Cree)脑炎 7 | X | ||||
家族性冻疮样狼疮(FLE) 8 | X | X |
1.TREX1的突变可能与该表中未包含的其他表型有关[ Rice et al 2015 ]。
2.所述杂合 ADAR p.Gly1007Arg变体,其已经显示出导致常染色体显性AGS [ Rice等2012 ],先前在两个个体与DSH证明肌张力障碍和颅内钙化神经变性描述东条等人2006,Kondo等人2008 ] 。
5.Richards等人[2007]已证明TREX1的C末端突变会引起RVCL,RVCL是常染色体显性遗传的微血管内皮病,与视网膜血管病,偏头痛,雷诺现象,中风和痴呆症相关,并在中年发病。这一发现提出了未经证实的可能性,即由TREX1致病性变异引起的AGS儿童的杂合父母,至少是那些在基因C端具有变异的儿童,有可能患上RVCL。
6.Lee-Kirsch等人[2007b]在417例SLE患者中的9例中发现了TREX1中的杂合致病变异。这一发现增加了由TREX1突变引起的AGS患者(以及其杂合的父母)可能会出现类似于SLE的体征和症状的风险。
7.关联分析和CSFIFN-α浓度的测定表明AGS和Cree脑炎是等位基因[ Crow et al 2003 ]。这证实,当分子遗传学检测结果显示:与克里脑炎所有的孩子都是纯合子的p.Arg164Ter 致病变种在TREX1。
8.Rice等人[2007年a]描述的杂合 TREX1 致病变种在受影响的与冻疮狼疮一个家族的成员; Lee-Kirsch等人[2007a]随后描述了第二种独特的致病变体。已经描述了其他致病变体。FLE也可能由SAMHD1中的杂合病原体变异引起[ Ravenscroft等,2011 ]。
除本综述中讨论的表型外,没有讨论与致病变异相关联的RNASEH2A或RNASEH2C的表型。
鉴别诊断
基底神经节的钙化是在许多疾病中观察到的非特异性发现。但是,在早发性脑病的情况下,要考虑的条件包括:
- TORCH先天的感染是鉴别中最常见的情况,也是最重要的排除因素,因为误诊会导致错误的复发风险咨询。
注意:在鉴别诊断中也应考虑其他先天的感染,例如与寨卡病毒和艾滋病毒相关的感染。
- 小头颅-颅内钙化综合征(MICS)。鉴于早发性Aicardi-Goutières综合征(AGS)病例的表型 (见临床特征Clinical Characteristics) [Reardon et al 1994],大多数MICS病例实际上是AGS(见Nomenclature)。但是,许多其他表型与新生儿颅内钙化有关[Knoblauch et al 2003, Gardner et al 2005 ]。因此,该表型无疑代表了一组异质的疾病(请参阅Nomenclature)。
- 带状钙化性多微子尿症(BLC-PMG;假性Torch综合征)(OMIM251290) [Abdel-Salam et al 2008, Briggs et al 2008]显示与AGS的放射学和临床重叠,表明颅内钙化和小头畸形和明显的精神运动障碍癫痫。可以通过观察多菌胞(在AGS中从未报道过)和在OCLN中鉴定双等位基因的 致病变异来区分该病[ O'Driscoll et al 2010]。
- 至少从表面上看,具有额颞叶白质改变和囊肿的AGS的MRI扫描结果可与Alexander病, 合并皮质下囊肿的大脑白质脑病/Alexander disease,megalencephalic leukoencephalopathy with subcortical cysts和儿童中枢神经系统低髓鞘/白质消失的共济失调/childhood ataxia with central nervous system hypomyelination/vanishing white matter disease混淆。早年的白质髓鞘过少程度也促使某些人考虑使用 Pelizeaus-Merzbacher 病。一般而言,在无法解释的白质脑病的鉴别诊断中应考虑使用AGS。该临床点特别重要,因为颅内钙化并不总是能在MRI上识别出来,MRI是大多数医疗机构采用的初始成像方式。
- 经典的Cockayne综合征 (CS型1)/Classic Cockayne syndrome是具有小脑钙化的白质营养不良,其特征在于其独特的面部特征,侏儒症,神经性耳聋,白内障,视网膜营养不良和皮肤光敏性。遗传是常染色体隐性遗传。
- 新生儿红斑狼疮。 Prendiville et al [2003]描述了新生儿红斑狼疮患儿的基底节钙化和斑块状白质减弱,令人联想到AGS的影像学发现。这些儿童表现出广泛的红斑性皮肤病变,与AGS中看到的冻疮病变不同。作者报告了这些病例的神经系统正常结果。
- Hoyeraal Hreidarsson综合征是先天性角化不全/dyskeratosis congenita的一种严重形式,由DKC1(X-linked)或TINF2(常染色体显性遗传)突变引起。生命的最初几个月出现Hoyeraal Hreidarsson综合征,伴小头畸形,小脑发育不全和脑钙化。受影响的男性发展出持续存在的全血细胞减少症(与某些患有AGS的个体所见的血小板减少症相反,后者通常会在生命的最初几周内消退)。
- 线粒体细胞病变,包括 Leigh syndrome和家族性线粒体脑病,并伴有脑钙化, Samson et al [1994]描述。另请参阅线粒体疾病概述/Mitochondrial Disorders Overview。
- 3-羟基异丁酸尿酸(OMIM 236795). Chitayat et al [1992]描述了单卵双生双胞胎,生于无血缘父母,具有变形的 面部特征,小头畸形,迁移性脑部疾病和先天的脑钙化。
- Blau等人[2003]描述了三名患有小头畸形,严重智力障碍和运动障碍,运动障碍,痉挛和偶尔发作的患者,这些患者的脑脊液中新蝶呤和生物蝶呤的浓度极高,而脑脊液中的5-甲基四氢叶酸浓度较低。尽管据报道具有AGS,但它们并未显示出CSF淋巴细胞增多或IFN-α浓度升高。因此,这些个体可能在患有脑病和颅内钙化的婴儿组中患有不确定的综合征。然而,现在已知在具有经分子证实的AGS的个体中可以观察到相似的蝶呤谱[ Rice等人2007b ]。
- 与钙化和囊肿Cerebroretinal微血管病 (CRMCC;外套加)(OMIM 612199)是造成双等位基因致病变种中CTC1 [乌鸦等人2004年b,Linnankivi等人,2006,Anderson等人2012,利文斯顿等人2014B ]。
- 脑白质脑病,脑钙化和囊肿(Labrune综合征)(OMIM 614561)
管理
初步诊断后的评估
为了确定诊断为心动过速-古特雷斯综合征(AGS)的个体的疾病程度和需求,建议进行以下评估:
- 发展评估
- 评估喂养和营养状况
- 眼科检查
- 如果怀疑有脑电图评估癫痫发作
- 咨询临床遗传学家和/或遗传咨询师
表现治疗
以下是适当的:
- 胸部物理疗法和大力治疗呼吸系统并发症
- 注意饮食和喂养方法,以确保摄入足够的热量
- 使用标准方案管理癫痫发作
监视
监视包括以下内容:
- 监测新生儿期尿崩症的体征
- 至少在生命的最初几年评估青光眼
- 监测脊柱侧弯的发展
- 监测胰岛素依赖型糖尿病和甲状腺功能减退的体征
评估亲戚处于危险中
有关为遗传咨询目的而对高危亲戚进行测试的相关问题,请参见遗传咨询。
正在调查的疗法
正在进行免疫抑制剂在AGS治疗中的研究[ Crow&Rehwinkel 2009,Crow et al 2014 ]。
在美国搜索ClinicalTrials.gov,在欧洲搜索欧盟临床试验注册,以获取有关多种疾病和状况的临床研究信息。
其他
皮质类固醇可以降低干扰素的脑脊液浓度[PG Barth 2003,个人交流];这种治疗的临床益处尚未得到证实。
注意:颅内大血管疾病与SAMHD1中双等位基因致病性变异相关的描述提出了有关此类个体管理的重要问题。所描述的闭塞性和动脉瘤性动脉病可以接受治疗(前者血运重建,后者卷曲或夹闭)。此外,动脉病的可能炎症基础表明免疫抑制可能在管理中起作用。一个关键问题是在临床表现时炎症性疾病是否活跃,或者所观察到的动脉异常是否代表了目前静止的炎症性过程的最终结果。
由于缺乏证据,目前无法就这些问题做出明确的陈述。但是,存在干预的潜力,并且可以说,某些人(例如精神运动问题较少的人)应该采取这种干预措施,并且应该仅对颅底动脉血管进行仔细检查,才应积极筛查颅内动脉病变。常规MRI可以看到大脑。Ramesh等人[2010]描述,所有受感染的个体中均存在冻疮样皮损,这可能是因为它们的存在预示着颅内血管病变的风险增加。
遗传咨询
遗传咨询
遗传咨询是为个人和家庭提供有关遗传疾病的性质,遗传和影响的信息的过程,以帮助他们做出明智的医疗和个人决定。下一节讨论遗传风险评估以及家族史和遗传测试的使用,以阐明家族成员的遗传状况。本部分的目的不是解决个人可能面临的所有个人,文化或道德问题,或替代与遗传学专业人员的咨询。—ED。
继承方式
RNASEH2A,RNASEH2B,RNASEH2C和SAMHD1 -相关Aicardi-Goutières综合征(AGS)是在遗传的常染色体隐性方式。
TREX1和ADAR相关的AGS可以以常染色体隐性或常染色体显性方式遗传,具体取决于特定的致病变异。
IFIH1相关性Aicardi-Goutières综合征(AGS)以常染色体显性方式遗传。
常染色体隐性遗传
对家庭成员的风险
先证者的父母
- 患病儿童的父母是致病杂合子(即,一种与AGS相关的致病变体的携带者)。
- 杂合子(携带者)没有发生AGS的风险;然而,Lee-Kirsch等人[2007b]和Richards等人[2007]的发现表明,杂合子患上迟发性系统性红斑狼疮(SLE)或伴有脑白质营养不良(RVCL)的视网膜血管病变的风险可能更高。有关的特定基因和病原体变异的信息(请参阅“遗传相关疾病”)。
先证者的同胞
- 受孕时,受影响个体的每个同胞都有25%的机会发病风险,有50%的机会成为无症状携带者,有25%的机会不受影响也不是携带者。
- 杂合子(携带者)没有发生AGS的风险;然而,取决于涉及的特定基因和病原体变异,杂合子患上迟发性系统性红斑狼疮(SLE)或伴有脑白质营养不良的视网膜血管病变的风险可能增加(参见遗传相关疾病)。
先证者的后代。大多数患有AGS的人不会繁殖。
其他家庭成员。先证者父母的每个同胞都有作为AGS相关病原体携带者的50%风险。
携带者(杂合子)检测
对高危亲属进行携带者检测需要事先鉴定该家族中与AGS相关的致病变异。
常染色体显性遗传–对家庭成员的风险
先证者的父母
- 迄今为止,大多数具有常染色体显性AGS的先证者由于新发生 TREX1,ADAR或IFIH1 致病性变异而患有该疾病[ Rice等,2007b;Haaxma等,2010;Rice等,2012;Abe等,2014 ]。
在致病变种的垂直传播TREX1(p.Asp18Asn; Abe等[2013]和ADAR(甘氨酸1007; Rice等人[2012],Livingston等人[2014a]已被报道。
- 建议用分子遗传学检测来评价具有明显新发生致病变异的先证者的父母。
- 如果在任何一方父母的白细胞DNA中均无法检测到在先证者中发现的病原体变异,则该先证者极有可能具有新发生的病原体变异。另一个可能的解释是亲本中的种系镶嵌(尽管理论上可能,没有报道种系镶嵌的实例)。
先证者的同胞。先证者同胞的风险取决于先证者父母的遗传状况:
- 如果一个家长先证者的影响,风险的同胞是50%。
- 如果先证者中发现的TREX1,ADAR或IFIH1 致病变体在任一亲本的白细胞DNA中均无法检测到,则由于以下原因,同胞患病的风险可能略高于一般人群(尽管仍<1%)父母种系镶嵌的理论可能性。
先证者的后代。具有常染色体显性遗传性AGS的个体的每个孩子都有50%的机会遗传AGS相关的致病变异;但是,大多数患有AGS的人不会繁殖。
其他家庭成员。对其他家庭成员的风险取决于先证者父母的状况:如果父母受到影响,则他或她的家庭成员可能处于危险之中。
相关的遗传咨询问题
家庭计划
DNA库是DNA(通常从白细胞中提取)的存储,以备将来使用。由于测试方法和我们对基因,等位基因变异和疾病的理解将来可能会改善,因此应考虑到受影响个体的库DNA 。
产前检测和植入前基因检测
一旦在受影响的家庭成员中确定了致病变体,就可以对风险较高的妊娠进行产前检查,并对AGS进行植入前基因检查。
Resources
GeneReviews staff has selected the following disease-specific and/or umbrellasupport organizations and/or registries for the benefit of individuals with this disorderand their families. GeneReviews is not responsible for the information provided by otherorganizations. For information on selection criteria, click here.
- International Aicardi-Goutières Syndrome Association (IAGSA)ItalyEmail: associazione.iagsa@tiscali.it; iagsa@libero.it
- National Library of Medicine Genetics Home Reference
- United Leukodystrophy Foundation (ULF)224 North Second StreetSuite 2DeKalb IL 60115Phone: 800-728-5483 (toll-free); 815-748-3211Fax: 815-748-0844Email: office@ulf.org
- Myelin Disorders Bioregistry ProjectEmail: myelindisorders@cnmc.org
分子遗传
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Aicardi-Goutieres 综合征: 基因和数据库
Table B.
OMIM Entries for Aicardi-Goutieres 综合征 (View All in OMIM)
| 146920 | 腺苷脱氨酶, RNA-特异性C; ADAR |
| 225750 | 艾卡迪-古铁雷斯综合征 1; AGS1 |
| 606034 | 核糖核酸酶 H2, 亚单位 A; RNASEH2A |
| 606609 | 3-PRIME @修复 核酸外切酶 1; TREX1 |
| 606754 | 含SAM 结构域- 和 HD 结构域 蛋白++ 1; SAMHD1 |
| 606951 | 干扰素诱导的螺旋酶C结构域蛋白 1; IFIH1 |
| 610181 | 艾卡迪-古铁雷斯综合征 2; AGS2 |
| 610326 | 核糖核酸酶 H2, 亚单位B; RNASEH2B |
| 610329 | 艾卡迪-古铁雷斯综合征 3; AGS3 |
| 610330 | 核糖核酸酶 H2, 亚单位 C; RNASEH2C |
| 610333 | 艾卡迪-古铁雷斯综合征 4; AGS4 |
| 612952 | 艾卡迪-古铁雷斯综合征 5; AGS5 |
| 615010 | 艾卡迪-古铁雷斯综合征 6; AGS6 |
| 615846 | 艾卡迪-古铁雷斯综合征 7; AGS7 |
ADAR
基因结构.ADAR单拷贝含16-外显子的 基因 t编码2个主要的 异型体 ,在哺乳动物内持续表达: 一种截短的变异 (p110 NP_001020278.1) 编码15个外显子 (NM_001025107.2) 和转录变体亚型 NM_001111.4诱导编码的一个 IFN包含蛋白质全长 (p150 NP_001102.2) .基因和蛋白的详细总结 见 Table A, Gene.
致病变异. ADAR中的大多数致病性变体导致 常染色体隐性遗传 病。多种错义,无义 nonsense, 移码,和 剪接位点 致病变异已被报告. 在欧洲血统的人中很常见这种 错义 变异, p.Pro193Ala。 一种显性遗传的变异, p.Gly1007Arg 被报告[Rice et al 2012, Livingston et al 2014a].
Table 4.
Selected ADAR Pathogenic Variants
| DNA 核苷酸改变 | 预测蛋白改变 | 序列信息 |
|---|---|---|
| c.577C>G | p.Pro193Ala | NM_001111 NP_001102 |
| c.3019G>A | p.Gly1007Arg |
Variants listed in the table have been provided by the author. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen
- .hgvs.org ). See Quick Reference for an explanation of nomenclature.
正常 基因产物. DRADA, 双链 RNA-特异性 腺苷脱氨酶,是一种具有C末端脱氨酶催化 结构域的模块化蛋白质, 三个位于中心的双链RNA结合域(dsRBDs)和一个或两个N端Z-DNA结合域;与p110相比,人类DRADA的p150亚型具有额外的295个N端氨基酸,包含核输出信号和额外的Z-DNA/Z-RNA结合域.
异常 基因产物. ADAR中的致病性变体被认为是由于蛋白质活性丧失而导致AGS;导致疾病的确切机制尚不清楚.
IFIH1
基因结构.IFIH1 有16 外显子. 关于 基因 和蛋白的详细总结,见 Table A, Gene.
致病变异. 至今所有与AGS相关的致病变异均为 错义 变异,导致显性遗传病.
正常 基因产物.IFIH1编码干扰素诱导的含螺旋酶C 结构域-蛋白质1,一种胞质双链RNA受体。
异常 基因产物. 与AGS编码相关的IHIF1致病性 错义变异 导致I型干扰素信号上调的变体 [Rice et al 2014].
RNASEH2A
基因结构.RNASEH2A有8个外显子. 关于 基因 和蛋白的详细总结,见 Table A, Gene.
致病变异. 已经报道的在 RNASEH2A 主要致病变异为 错义; 剪接 和移码 [Crow et al 2006b, Rice et al 2007b] (see Table 5).
Table 5.
Selected RNASEH2A Pathogenic Variants
| DNA 核苷酸变异 1 | 预测的蛋白质改变 | 序列信息 |
|---|---|---|
| c.69G>A 2 | p.Val23= | NM_006397 NP_006388 |
| c.75C>T 2 | p.Arg25= | |
| c.109G>A | p.Gly37Ser | |
| c.207_208insG | p.Thr69AspfsTer50 | |
| c.322C>T | p.Arg108Trp | |
| c.556C>T | p.Arg186Trp | |
| c.690C>A | p.Phe231Leu | |
| c.704G>A | p.Arg236Gln | |
| c.716_717dupGC | p.Thr239AlafsTer77 | |
| c.719C>T | p.Thr241Met | |
| c.872G>A | p.Arg292His |
Variants listed in the table have been provided by the author. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen
- .hgvs.org ). See Quick Reference for an explanation of nomenclature.- 1.
每个 等位基因 的频率低于 1%.
- 2.
RNASEH2A的2个同义变异 (c.69G>A and c.75C>T) 也是致病的,可以导致 RNA 剪接 位点改变[Rice et al 2013b].
正常 基因产物.核糖核酸酶H2A编码核糖核酸酶H2亚单位A,由299个氨基酸组成。核糖核酸酶H(RNASEH)酶从RNA:DNA双链体内切核苷酸。RNASEH2被认为在DNA复制过程中起到去除滞后链冈崎片段RNA引物的作用,以及从DNA:DNA双链体中去除单个核糖核苷酸的作用。然而,人类RNASEH2复合物在AGS中的确切生物学功能尚不确定。
异常 基因产物. 见 RNASEH2B,Abnormal gene product.
RNASEH2B
基因结构.RNASEH2B 有11个外显子,编码 308氨基酸的蛋白质. 关于 基因 和蛋白的详细总结,见 Table A, Gene.
致病变异. 至今已确定的 RNASEH2B致病变异均为 错义突变 [Crow et al 2006b, Rice et al 2007b] (see Table 6).
在 受累的 个体检测出的致病变异频率为:
- p.Ala177Thr (62%)
- p.Thr163Ile (7%)
- p.Val185Gly (7%)
- c.136+1delG (4%)
- 其他等位基因变异 (<2%) [Rice et al 2007b]
Table 6.
Selected RNASEH2B Pathogenic Variants
| DNA 核苷酸改变 | 预测的蛋白质改变 | 序列信息 |
|---|---|---|
| c.64+1G>A | -- | NM_024570 NP_078846 |
| c.136+1delG | -- | |
| c.244+1G>T | -- | |
| c.436+1G>T | -- | |
| c.510+1G>A | -- | |
| c.128C>A | p.Pro43His | |
| c.132T>A | p.Cys44Ter | |
| c.172C>T | p.Gln58Ter | |
| c.179T>G | p.Leu60Arg | |
| c.218G>T | p.Trp73Leu | |
| c.247G>A | p.Gly83Ser | |
| c.257A>G | p.His86Arg | |
| c.412C>T | p.Leu138Phe | |
| c.476G>T | p.Ser159Ile | |
| c.485A>C | p.Lys162Thr | |
| c.488C>T | p.Thr163Ile | |
| c.529G>A | p.Ala177Thr | |
| c.547C>A | p.Val183Met | |
| c.554T>G | p.Val185Gly | |
| c.655T>C | p.Tyr219His |
Variants listed in the table have been provided by the author. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen
- .hgvs.org ). See Quick Reference for an explanation of nomenclature.
正常 基因产物. 核糖核酸酶H2亚单位B蛋白在人类核糖核酸酶H2复合物中的确切功能尚不清楚。
异常 基因产物. 编码核糖核酸酶H2复合物三个亚基中任何一个亚基的基因中的致病性变异都被认为是由于酶功能丧失而导致AGS。
RNASEH2C
基因结构.RNASEH2C 是有4个-外显子 的基因编码一个含 164氨基酸的蛋白.关于 基因 和蛋白的详细总结,见 Table A, Gene.
致病变异. 至今报告的所有 RNASEH2C致病变异均为 错义变异 [Crow et al 2006b, Rice et al 2007b] (see Table 7).
确定受累的个体等位基因频率为:
- p.Arg69Trp (72%)
- 其他 (仅见单个家庭)
Table 7.
Selected RNASEH2C Pathogenic Variants
| DNA 核苷酸改变 | 预测蛋白质改变 | 序列信息 |
|---|---|---|
| c.38G>A | p.Arg13His | NM_032193 NP_115569 |
| c.205C>T | p.Arg69Trp | |
| c.227C>T | p.Pro76Leu | |
| c.412C>T | p.Pro138Leu | |
| c.428A>T | p.Lys143Ile | |
| c.451C>T | p.Pro151Ser |
Variants listed in the table have been provided by the author. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen
- .hgvs.org ). See Quick Reference for an explanation of nomenclature.
正常 基因产物. 核糖核酸酶H2亚单位C(RNASEH2C)在RNASEH2复合物中的功能尚不清楚。
异常 基因产物. 见 RNASEH2B,异常基因产物.
SAMHD1
基因结构.SAMHD1有 16 个外显子. For a detailed summary of 基因 and protein information, see Table A, Gene.
Pathogenic variants. The majority of pathogenic variants are 错义, 剪接位点, nonsense, or frameshift variants [Rice et al 2009].
Three large deletions plus a small 缺失 have also been identified. A large deletion of 外显子 1 is common among individuals of Ashkenazi Jewish descent [Crow et al 2015].
Table 8.
Selected SAMHD1 Pathogenic Variants
| DNA Nucleotide Change (Alias 1) | Predicted Protein Change | Reference Sequences |
|---|---|---|
| c.359_370del12 | p.Asp120_His123del | NM_015474 NP_056289 |
| c.368A>C | p.His123Pro | |
| c.427C>T | p.Arg143Cys | |
| c.428G>A | p.Arg143His | |
| c.433C>T | p.Arg145Ter | |
| c.434G>A | p.Arg145Gln | |
| c.445C>T | p.Gln149Ter | |
| c.602T>A | p.Ile201Asn | |
| c.625G>A | p.Gly209Ser | |
| c.649_650insG | p.Phe217CysTer2 | |
| c.760A>G | p.Met254Val | |
| c.1106T>C | p.Leu369Ser | |
| c.1153A>G | p.Met385Val | |
| c.1324C>T | p.Arg442Ter | |
| c.1411-2A>G | (splice acceptor) | |
| c.1503+1G>T | (splice donor) | |
| c.1642C>T | p.Gln548Ter | |
| c.1609-1G>C | (splice acceptor) | |
| (exons 12-16del) | -- | |
| (8984bp promoter+ex1del) | -- | |
| (exons 1-13del) | -- |
Variants listed in the table have been provided by the author. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen
- .hgvs.org ). See Quick Reference for an explanation of nomenclature.- 1.
Variant designation that does not conform to current naming conventions
正常 基因产物. SAMHD1是由SAM组成的 626氨基酸蛋白质 结构域 和一个HD域;该蛋白作为dNTP三磷酸水解酶发挥作用 [Goldstone et al 2011].
异常 基因产物. SAMHD1中的致病性变异被认为是由于蛋白质的错误定位或HD结构域的丢失而导致AGS;该蛋白作为具有蛋白质功能的dNTP三磷酸水解酶起作用
TREX1 (AGS1)
基因结构.TREX1 只有一个 外显子. 关于 基因 和蛋白的详细总结,见 Table A, Gene.
注意: 在数据库中,TREX1 和一个重叠的 基因-ATRIP存在大量混乱的信息 。TREX1和ATRIP是编码不同蛋白质的不同基因;它们之间没有相关性 [Yang et al 2007].
致病变异. 在TREX1中终止密码变异、缺失和插入很常见,但最普遍致病性变异 是一个错义 影响TREX1蛋白(3'修复核酸外切酶1)二聚化的变体(p.Arg114His),可能是一种功能性无效 等位基因。 致病性变种p.Arg114His在北欧人群中尤其常见
受累个体 几乎总是这个基因的纯合或复合杂合致病变异。然而, 伴有典型 AGS的患儿有TREX1新发生/ de novo 杂合的 致病性变异 . [Rice et al 2007a, Haaxma et al 2010, Abe et al 2014] (see Table 9).
受累的 个体 等位基因频率[Rice et al 2007b]:
- p.Arg114His (50%)
- c.58_59insG (1%)
- 其他(<1%)
Table 9.
Selected TREX1 Pathogenic Variants
| DNA Nucleotide Change | Predicted Protein Change | Reference Sequences |
|---|---|---|
| c.52G>A | p.Asp18Asn | AAK07616 AF319569 |
| c.58_59insG | p.Glu20GlyfsTer81 | |
| c.212_213dupTG | p.Ala72Trpfster16 | |
| c.341G>A | p.Arg114His | |
| c.365T>C | p.Val122Ala | |
| c.366_368dupGGC | p.Ala123dup | |
| c.397delC | p.Leu133CysfsTer26 | |
| c.393_408dup16 | p.Glu137ProfsTer23 | |
| c.490C>T | p.Arg164Ter | |
| c.500delG | p.Ser166ThrfsTer12 | |
| c.598G>T | p.Asp200Asn | |
| c.600_601insGAT | p.Asp200dup | |
| c.602T>A | p.Val201Asp | |
| c.609_662dup54 | p.Leu204_Ala221dup | |
| c.625_628dupCAGT | p.Trp210SerfsTer31 | |
| c.868_885del18 | p.Pro289_Ala294del | |
| c.907A>C | p.Thr303Pro |
Variants listed in the table have been provided by the author. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen
- .hgvs.org ). See Quick Reference for an explanation of nomenclature.
正常 基因产物.TREX1 为单个-外显子 基因,编码含 314氨基酸的蛋白质.
TREX1蛋白代表主要的3'→哺乳动物细胞中测定的5'DNA外切酶活性。该蛋白质有三个保守的序列基序,称为外显子I、II和III。这些基序包含四个保守的酸性残基,参与催化所需的二价金属离子的配位。另外,这个蛋白含有大约75的氨基酸的 C端 结构域 , 它可能参与蛋白质的亚细胞定位,以及可能参与与其他蛋白质相互作用的多聚脯氨酸基序。TREX1似乎在单链DNA的处理中发挥作用,单链DNA可能在S期作为正常复制中间产物产生 [Yang et al 2007], 或者衍生于一个逆反元素. [Stetson et al 2008].
异常基因产物.TREX1 致病性变异最常见的是一个 错义 变异 (p.Arg114His),这影响TREX1蛋白(3'修复核酸外切酶1)的二聚化,可能是一种功能性蛋白无效 等位基因, 其他报道的移码变异体也是如此。
References
Literature Cited
- Abdel-Salam GM, Zaki MS, Saleem SN, Gaber KR. Microcephaly, malformation of brain development and intracranial calcification in sibs: pseudo-TORCH or a new syndrome. Am J Med Genet A. 2008;146A:2929 - 36. [PubMed: 18925673]
- Abe J, Izawa K, Nishikomori R, Awaya T, Kawai T, Yasumi T, Hiragi N, Hiragi T, Ohshima Y, Heike T. Heterozygous TREX1 p.Asp18Asn mutation can cause variable neurological symptoms in a family with Aicardi-Goutieres syndrome/familial chilblain lupus. Rheumatology (Oxford). 2013;52:406 - 8. [PubMed: 22829693]
- Abe J, Nakamura K, Nishikomori R, Kato M, Mitsuiki N, Izawa K, Awaya T, Kawai T, Yasumi T, Toyoshima I, Hasegawa K, Ohshima Y, Hiragi T, Sasahara Y, Suzuki Y, Kikuchi M, Osaka H, Ohya T, Ninomiya S, Fujikawa S, Akasaka M, Iwata N, Kawakita A, Funatsuka M, Shintaku H, Ohara O, Ichinose H, Heike T. A nationwide survey of Aicardi-Goutieres syndrome patients identifies a strong association between dominant TREX1 mutations and chilblain lesions: Japanese cohort study. Rheumatology (Oxford). 2014;53:448 - 58. [PubMed: 24300241]
- Aicardi J, Goutières F. A progressive familial encephalopathy in infancy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis. Ann Neurol. 1984;15:49 - 54. [PubMed: 6712192]
- Aicardi J, Goutières F. Systemic lupus erythematosus or Aicardi-Goutieres syndrome? Neuropediatrics. 2000;31:113. [PubMed: 10963096]
- Anderson BH, Kasher PR, Mayer J, Szynkiewicz M, Jenkinson EM, Bhaskar SS, Urquhart JE, Daly SB, Dickerson JE, O'Sullivan J, Leibundgut EO, Muter J, Abdel-Salem GM, Babul-Hirji R, Baxter P, Berger A, Bonafé L, Brunstom-Hernandez JE, Buckard JA, Chitayat D, Chong WK, Cordelli DM, Ferreira P, Fluss J, Forrest EH, Franzoni E, Garone C, Hammans SR, Houge G, Hughes I, Jacquemont S, Jeannet PY, Jefferson RJ, Kumar R, Kutschke G, Lundberg S, Lourenço CM, Mehta R, Naidu S, Nischal KK, Nunes L, Ounap K, Philippart M, Prabhakar P, Risen SR, Schiffmann R, Soh C, Stephenson JB, Stewart H, Stone J, Tolmie JL, van der Knaap MS, Vieira JP, Vilain CN, Wakeling EL, Wermenbol V, Whitney A, Lovell SC, Meyer S, Livingston JH, Baerlocher GM, Black GC, Rice GI, Crow YJ. Mutations in CTC1, encoding conserved telomere maintenance component 1, cause Coats plus. Nat Genet. 2012;44:338 - 42. [PubMed: 22267198]
- Barth PG (2002) The neuropathology of Aicardi-Goutieres syndrome. Eur J Paediatr Neurol. 6 Suppl A:A27-31. [PubMed: 12365358]
- Blau N, Bonafe L, Krageloh-Mann I, Thony B, Kierat L, Hausler M, Ramaekers V. Cerebrospinal fluid pterins and folates in Aicardi-Goutieres syndrome: a new phenotype. Neurology. 2003;61:642 - 7. [PubMed: 12963755]
- Briggs TA, Wolf NI, D'Arrigo S, Ebinger F, Harting I, Dobyns WB, Livingston JH, Rice GI, Crooks D, Rowland-Hill CA, Squier W, Stoodley N, Pilz DT, Crow YJ. Band-like intracranial calcification with simplified gyration and polymicrogyria: a distinct "pseudo-TORCH" phenotype. Am J Med Genet A. 2008;146A:3173 - 80. [PubMed: 19012351]
- Chitayat D, Meagher-Villemure K, Mamer OA, O'Gorman A, Hoar DI, Silver K, Scriver CR. Brain dysgenesis and congenital intracerebral calcification associated with 3-hydroxyisobutyric aciduria. J Pediatr. 1992;121:86 - 9. [PubMed: 1625099]
- Crow YJ, Black DN, Ali M, Bond J, Jackson AP, Lefson M, Michaud J, Roberts E, Stephenson JB, Woods CG, Lebon P. Cree encephalitis is allelic with Aicardi-Goutieres syndrome: implications for the pathogenesis of disorders of interferon alpha metabolism. J Med Genet. 2003;40:183 - 7. [PMC free article: PMC1735395] [PubMed: 12624136]
- Crow YJ, Chase DS, Lowenstein Schmidt J, Szynkiewicz M, Forte GM, Gornall HL, Oojageer A, Anderson B, Pizzino A, Helman G, Abdel-Hamid MS, Abdel-Salam GM, Ackroyd S, Aeby A, Agosta G, Albin C, Allon-Shalev S, Arellano M, Ariaudo G, Aswani V, Babul-Hirji R, Baildam EM, Bahi-Buisson N, Bailey KM, Barnerias C, Barth M, Battini R, Beresford MW, Bernard G, Bianchi M, Billette de Villemeur T, Blair EM, Bloom M, Burlina AB, Carpanelli ML, Carvalho DR, Castro-Gago M, Cavallini A, Cereda C, Chandler KE, Chitayat DA, Collins AE, Sierra Corcoles C, Cordeiro NJ, Crichiutti G, Dabydeen L, Dale RC, D'Arrigo S, De Goede CG, De Laet C, De Waele LM, Denzler I, Desguerre I, Devriendt K, Di Rocco M, Fahey MC, Fazzi E, Ferrie CD, Figueiredo A, Gener B, Goizet C, Gowrinathan NR, Gowrishankar K, Hanrahan D, Isidor B, Kara B, Khan N, King MD, Kirk EP, Kumar R, Lagae L, Landrieu P, Lauffer H, Laugel V, La Piana R, Lim MJ, Lin JP, Linnankivi T, Mackay MT, Marom DR, Marques Lourenço C, McKee SA, Moroni I, Morton JE, Moutard ML, Murray K, Nabbout R, Nampoothiri S, Nunez-Enamorado N, Oades PJ, Olivieri I, Ostergaard JR, Pérez-Dueñas B, Prendiville JS, Ramesh V, Rasmussen M, Régal L, Ricci F, Rio M, Rodriguez D, Roubertie A, Salvatici E, Segers KA, Sinha GP, Soler D, Spiegel R, Stödberg TI, Straussberg R, Swoboda KJ, Suri M, Tacke U, Tan TY, te Water Naude J, Wee Teik K, Thomas MM, Till M, Tonduti D, Valente EM, Van Coster RN, van der Knaap MS, Vassallo G, Vijzelaar R, Vogt J, Wallace GB, Wassmer E, Webb HJ, Whitehouse WP, Whitney RN, Zaki MS, Zuberi SM, Livingston JH, Rozenberg F, Lebon P, Vanderver A, Orcesi S, Rice GI. Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am J Med Genet A. 2015;2015;167A:296 - 312. [PMC free article: PMC4382202] [PubMed: 25604658]
- Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, Black DN, van Bokhoven H, Brunner HG, Hamel BC, Corry PC, Cowan FM, Frints SG, Klepper J, Livingston JH, Lynch SA, Massey RF, Meritet JF, Michaud JL, Ponsot G, Voit T, Lebon P, Bonthron DT, Jackson AP, Barnes DE, Lindahl T. Mutations in the gene encoding the 3'-5' DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nat Genet. 2006a;38:917 - 20. [PubMed: 16845398]
- Crow YJ, Leitch A, Hayward BE, Garner A, Parmar R, Griffith E, Ali M, Semple C, Aicardi J, Babul-Hirji R, Baumann C, Baxter P, Bertini E, Chandler KE, Chitayat D, Cau D, Dery C, Fazzi E, Goizet C, King MD, Klepper J, Lacombe D, Lanzi G, Lyall H, Martinez-Frias ML, Mathieu M, McKeown C, Monier A, Oade Y, Quarrell OW, Rittey CD, Rogers RC, Sanchis A, Stephenson JB, Tacke U, Till M, Tolmie JL, Tomlin P, Voit T, Weschke B, Woods CG, Lebon P, Bonthron DT, Ponting CP, Jackson AP. Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutieres syndrome and mimic congenital viral brain infection. Nat Genet. 2006b;38:910 - 6. [PubMed: 16845400]
- Crow YJ, Massey RF, Innes JR, Pairaudeau PW, Rowland Hill CA, Woods CG, Ali M, Livingston JH, Lebon P, Nischall K, McEntagart M, Hindocha N, Winter RM. Congenital glaucoma and brain stem atrophy as features of Aicardi-Goutieres syndrome. Am J Med Genet. 2004a;129A:303 - 7. [PubMed: 15326633]
- Crow YJ, McMenamin J, Haenggeli CA, Hadley DM, Tirupathi S, Treacy EP, Zuberi SM, Browne BH, Tolmie JL, Stephenson JB. Coats' plus: a progressive familial syndrome of bilateral Coats' disease, characteristic cerebral calcification, leukoencephalopathy, slow pre- and post-natal linear growth and defects of bone marrow and integument. Neuropediatrics. 2004b;35:10 - 9. [PubMed: 15002047]
- Crow YJ, Rehwinkel J. Aicardi-Goutieres syndrome and related phenotypes: linking nucleic acid metabolism with autoimmunity. Hum Mol Genet. 2009;18:R130 - 6. [PMC free article: PMC2758706] [PubMed: 19808788]
- Crow YJ, Vanderver A, Orcesi S, Kuijpers TW, Rice GI. Therapies in Aicardi-Goutières syndrome. Clin Exp Immunol. 2014;175:1 - 8. [PMC free article: PMC3898548] [PubMed: 23607857]
- Crow YJ, Zaki MS, Abdel-Hamid MS, Abdel-Salam G, Boespflug-Tanguy O, Cordeiro NJ, Gleeson JG, Gowrinathan NR, Laugel V, Renaldo F, Rodriguez D, Livingston JH, Rice GI. Mutations in ADAR1, IFIH1, and RNASEH2B presenting as spastic paraplegia. Neuropediatrics. 2014a;45:386 - 93. [PubMed: 25243380]
- Dale RC, Gornall H, Singh-Grewal D, Alcausin M, Rice GI, Crow YJ. Familial Aicardi-Goutières syndrome due to SAMHD1 mutations is associated with chronic arthropathy and contractures. Am J Med Genet A. 2010;152A:938 - 42. [PubMed: 20358604]
- Dale RC, Tang SP, Heckmatt JZ, Tatnall FM. Familial systemic lupus erythematosus and congenital infection-like syndrome. Neuropediatrics. 2000;31:155 - 8. [PubMed: 10963105]
- Desanges C, Lebon P, Bauman C, Vuillard E, Garel C, Cordesse A, Oury JF, Crow Y, Luton D. Elevated interferon-alpha in fetal blood in the prenatal diagnosis of Aicardi-Goutieres syndrome. Fetal Diagn Ther. 2006;21:153 - 5. [PubMed: 16354995]
- du Moulin M, Nürnberg P, Crow YJ, Rutsch F. Cerebral vasculopathy is a common feature in Aicardi-Goutieres syndrome associated with SAMHD1 mutations. Proc Natl Acad Sci U S A. 2011;108:E232 [PMC free article: PMC3127916] [PubMed: 21633013]
- Gardner RJ, Chow CW, Simpson I, Fink AM, Meagher SE, White SM. Severe fetal brain dysgenesis with focal calcification. Prenat Diagn. 2005;25:362 - 4. [PubMed: 15906425]
- Goldstone DC, Ennis-Adeniran V, Hedden JJ, Groom HC, Rice GI, Christodoulou E, Walker PA, Kelly G, Haire LF, Yap MW, de Carvalho LP, Stoye JP, Crow YJ, Taylor IA, Webb M. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature. 2011;480:379 - 82. [PubMed: 22056990]
- Goutières F, Aicardi J, Barth PG, Lebon P. Aicardi-Goutieres syndrome: an update and results of interferon-alpha studies. Ann Neurol. 1998;44:900 - 7. [PubMed: 9851434]
- Haaxma CA, Crow YJ, van Steensel MA, Lammens MM, Rice GI, Verbeek MM, Willemsen MA. A de novo p.Asp18Asn mutation in TREX1 in a patient with Aicardi-Goutières syndrome. Am J Med Genet A. 2010;152A:2612 - 7. [PubMed: 20799324]
- Knoblauch H, Tennstedt C, Brueck W, Hammer H, Vulliamy T, Dokal I, Lehmann R, Hanefeld F, Tinschert S. Two brothers with findings resembling congenital intrauterine infection-like syndrome (pseudo-TORCH syndrome). Am J Med Genet A. 2003;120A:261 - 5. [PubMed: 12833411]
- Kondo T, Suzuki T, Ito S, Kono M, Negoro T, Tomita Y. Dyschromatosis symmetrica hereditaria associated with neurological disorders. J Dermatol. 2008;35:662 - 6. [PubMed: 19017046]
- Kumar D, Rittey C, Cameron AH, Variend S. Recognizable inherited syndrome of progressive central nervous system degeneration and generalized intracranial calcification with overlapping phenotype of the syndrome of Aicardi and Goutieres. Am J Med Genet. 1998;75:508 - 15. [PubMed: 9489795]
- La Piana R, Uggetti C, Olivieri I, Tonduti D, Balottin U, Fazzi E, Orcesi S. Bilateral striatal necrosis in two subjects with Aicardi-Goutières syndrome due to mutations in ADAR1 (AGS6). Am J Med Genet A. 2014;164A:815 - 9. [PubMed: 24376015]
- Lanzi G, Fazzi E, D'Arrigo S. Aicardi-Goutieres syndrome: a description of 21 new cases and a comparison with the literature. Eur J Paediatr Neurol. 2002;6:A9鈥揂22. [PubMed: 12365365]
- Lanzi G, Fazzi E, D'Arrigo S, Orcesi S, Maraucci I, Uggetti C, Bertini E, Lebon P. The natural history of Aicardi-Goutieres syndrome: follow-up of 11 Italian patients. Neurology. 2005;64:1621 - 4. [PubMed: 15883328]
- Le Garrec M, Doret M, Pasquier JC, Till M, Lebon P, Buenerd A, Escalon J, Gaucherand P. Prenatal diagnosis of Aicardi-Goutieres syndrome. Prenat Diagn. 2005;25:28 - 30. [PubMed: 15662687]
- Lee-Kirsch MA, Chowdhury D, Harvey S, Gong M, Senenko L, Engel K, Pfeiffer C, Hollis T, Gahr M, Perrino FW, Lieberman J, Hubner N. A mutation in TREX1 that impairs susceptibility to granzyme A-mediated cell death underlies familial chilblain lupus. J Mol Med. 2007a;85:531 - 7. [PubMed: 17440703]
- Lee-Kirsch MA, Gong M, Chowdhury D, Senenko L, Engel K, Lee YA, de Silva U, Bailey SL, Witte T, Vyse TJ, Kere J, Pfeiffer C, Harvey S, Wong A, Koskenmies S, Hummel O, Rohde K, Schmidt RE, Dominiczak AF, Gahr M, Hollis T, Perrino FW, Lieberman J, Hubner N. Mutations in the gene encoding the 3'-5' DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat Genet. 2007b;39:1065 - 7. [PubMed: 17660818]
- Linnankivi T, Valanne L, Paetau A, Alafuzoff I, Hakumaki JM, Kivela T, Lonnqvist T, Makitie O, Paakkonen L, Vainionpaa L, Vanninen R, Herva R, Pihko H. Cerebroretinal microangiopathy with calcifications and cysts. Neurology. 2006;67:1437 - 43. [PubMed: 16943371]
- Livingston JH, Lin JP, Dale RC, Gill D, Brogan P, Munnich A, Kurian MA, Gonzalez-Martinez V, De Goede CG, Falconer A, Forte G, Jenkinson EM, Kasher PR, Szynkiewicz M, Rice GI, Crow YJ. A type I interferon signature identifies bilateral striatal necrosis due to mutations in ADAR1. J Med Genet. 2014a;51:76 - 82. [PubMed: 24262145]
- Livingston JH, Mayer J, Jenkinson E, Kasher P, Stivaros S, Berger A, Cordelli DM, Ferreira P, Jefferson R, Kutschke G, Lundberg S, Ounap K, Prabhakar P, Soh C, Stewart H, Stone J, van der Knaap MS, van Esch H, van Mol C, Wakeling E, Whitney A, Rice GI, Crow YJ. Leukoencephalopathy with calcifications and cysts: a purely neurological disorder distinct from Coats plus. Neuropediatrics. 2014b;45:175 - 82. [PubMed: 24407470]
- Livingston JH, Stivaros S, van der Knaap MS, Crow YJ. Recognizable phenotypes associated with intracranial calcification. Dev Med Child Neurol. 2013;55:46 - 57. [PubMed: 23121296]
- McEntagart M, Kamel H, Lebon P, King MD. Aicardi-Goutieres syndrome: an expanding phenotype. Neuropediatrics. 1998;29:163 - 7. [PubMed: 9706629]
- O'Driscoll MC, Daly SB, Urquhart JE, Black GC, Pilz DT, Brockmann K, McEntagart M, Abdel-Salam G, Zaki M, Wolf NI, Ladda RL, Sell S, D'Arrigo S, Squier W, Dobyns WB, Livingston JH, Crow YJ. Recessive mutations in the gene encoding the tight junction protein occludin cause band-like calcification with simplified gyration and polymicrogyria. Am J Hum Genet. 2010;87:354 - 64. [PMC free article: PMC2933344] [PubMed: 20727516]
- Prendiville JS, Cabral DA, Poskitt KJ, Au S, Sargent MA. Central nervous system involvement in neonatal lupus erythematosus. Pediatr Dermatol. 2003;20:60 - 7. [PubMed: 12558850]
- Ramantani G, Kohlhase J, Hertzberg C, Innes AM, Engel K, Hunger S, Borozdin W, Mah JK, Ungerath K, Walkenhorst H, Richardt HH, Buckard J, Bevot A, Siegel C, von Stülpnagel C, Ikonomidou C, Thomas K, Proud V, Niemann F, Wieczorek D, Häusler M, Niggemann P, Baltaci V, Conrad K, Lebon P, Lee-Kirsch MA. Expanding the phenotypic spectrum of lupus erythematosus in Aicardi-Goutières syndrome. Arthritis Rheum. 2010;62:1469 - 77. [PubMed: 20131292]
- Ramesh V, Bernardi B, Stafa A, Garone C, Franzoni E, Abinun M, Mitchell P, Mitra D, Friswell M, Nelson J, Shalev SA, Rice GI, Gornall H, Szynkiewicz M, Aymard F, Ganesan V, Prendiville J, Livingston JH, Crow YJ. Intracerebral large artery disease in Aicardi-Goutières syndrome implicates SAMHD1 in vascular homeostasis. Dev Med Child Neurol. 2010;52:725 - 32. [PubMed: 20653736]
- Ravenscroft JC, Suri M, Rice GI, Szynkiewicz M, Crow Y. Autosomal dominant inheritance of a heterozygous mutation in SAMHD1 causing familial chilblain lupus. Am J Med Genet A. 2011;155A:235 - 7. [PubMed: 21204240]
- Reardon W, Hockey A, Silberstein P, Kendall B, Farag TI, Swash M, Stevenson R, Baraitser M. Autosomal recessive congenital intrauterine infection-like syndrome of microcephaly, intracranial calcification, and CNS disease. Am J Med Genet. 1994;52:58 - 65. [PubMed: 7977464]
- Rice G, Newman WG, Dean J, Patrick T, Parmar R, Flintoff K, Robins P, Harvey S, Hollis T, O'Hara A, Herrick AL, Bowden AP, Perrino FW, Lindahl T, Barnes DE, Crow YJ. Heterozygous mutations in TREX1 cause familial chilblain lupus and dominant Aicardi-Goutieres syndrome. Am J Hum Genet. 2007a;80:811 - 5. [PMC free article: PMC1852703] [PubMed: 17357087]
- Rice G, Patrick T, Parmar R, Taylor CF, Aeby A, Aicardi J, Artuch R, Montalto SA, Bacino CA, Barroso B, Baxter P, Benko WS, Bergmann C, Bertini E, Biancheri R, Blair EM, Blau N, Bonthron DT, Briggs T, Brueton LA, Brunner HG, Burke CJ, Carr IM, Carvalho DR, Chandler KE, Christen HJ, Corry PC, Cowan FM, Cox H, D'Arrigo S, Dean J, De Laet C, De Praeter C, Dery C, Ferrie CD, Flintoff K, Frints SG, Garcia-Cazorla A, Gener B, Goizet C, Goutieres F, Green AJ, Guet A, Hamel BC, Hayward BE, Heiberg A, Hennekam RC, Husson M, Jackson AP, Jayatunga R, Jiang YH, Kant SG, Kao A, King MD, Kingston HM, Klepper J, van der Knaap MS, Kornberg AJ, Kotzot D, Kratzer W, Lacombe D, Lagae L, Landrieu PG, Lanzi G, Leitch A, Lim MJ, Livingston JH, Lourenco CM, Lyall EG, Lynch SA, Lyons MJ, Marom D, McClure JP, McWilliam R, Melancon SB, Mewasingh LD, Moutard ML, Nischal KK, Ostergaard JR, Prendiville J, Rasmussen M, Rogers RC, Roland D, Rosser EM, Rostasy K, Roubertie A, Sanchis A, Schiffmann R, Scholl-Burgi S, Seal S, Shalev SA, Corcoles CS, Sinha GP, Soler D, Spiegel R, Stephenson JB, Tacke U, Tan TY, Till M, Tolmie JL, Tomlin P, Vagnarelli F, Valente EM, Van Coster RN, Van der Aa N, Vanderver A, Vles JS, Voit T, Wassmer E, Weschke B, Whiteford ML, Willemsen MA, Zankl A, Zuberi SM, Orcesi S, Fazzi E, Lebon P, Crow YJ. Clinical and molecular phenotype of Aicardi-Goutieres syndrome. Am J Hum Genet. 2007b;81:713 - 25. [PMC free article: PMC2227922] [PubMed: 17846997]
- Rice GI, Bond J, Asipu A, Brunette RL, Manfield IW, Carr IM, Fuller JC, Jackson RM, Lamb T, Briggs TA, Ali M, Gornall H, Couthard LR, Aeby A, Attard-Montalto SP, Bertini E, Bodemer C, Brockmann K, Brueton LA, Corry PC, Desguerre I, Fazzi E, Cazorla AG, Gener B, Hamel BC, Heiberg A, Hunter M, van der Knaap MS, Kumar R, Lagae L, Landrieu PG, Lourenco CM, Marom D, McDermott MF, van der Merwe W, Orcesi S, Prendiville JS, Rasmussen M, Shalev SA, Soler DM, Shinawi M, Spiegel R, Tan TY, Vanderver A, Wakeling EL, Wassmer E, Whittaker E, Lebon P, Stetson DB, Bonthron DT, Crow YJ. Mutations involved in Aicardi-Goutières syndrome implicate SAMHD1 as regulator of the innate immune response. Nat Genet. 2009;41:829 - 32. [PMC free article: PMC4154505] [PubMed: 19525956]
- Rice GI, del Toro Duany Y, Jenkinson EM, Forte GM, Anderson BH, Ariaudo G, Bader-Meunier B, Baildam EM, Battini R, Beresford MW, Casarano M, Chouchane M, Cimaz R, Collins AE, Cordeiro NJ, Dale RC, Davidson JE, De Waele L, Desguerre I, Faivre L, Fazzi E, Isidor B, Lagae L, Latchman AR. Lebon P2, Li C, Livingston JH, Lourenço CM, Mancardi MM, Masurel-Paulet A, McInnes IB, Menezes MP, Mignot C, O'Sullivan J, Orcesi S, Picco PP, Riva E, Robinson RA, Rodriguez D, Salvatici E, Scott C, Szybowska M, Tolmie JL, Vanderver A, Vanhulle C, Vieira JP, Webb K, Whitney RN, Williams SG, Wolfe LA, Zuberi SM, Hur S, Crow YJ. Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat Genet. 2014;46:503 - 9. [PMC free article: PMC4004585] [PubMed: 24686847]
- Rice GI, Forte GM, Szynkiewicz M, Chase DS, Aeby A, Abdel-Hamid MS, Ackroyd S, Allcock R, Bailey KM, Balottin U, Barnerias C, Bernard G, Bodemer C, Botella MP, Cereda C, Chandler KE, Dabydeen L, Dale RC, De Laet C, De Goede CG, Del Toro M, Effat L, Enamorado NN, Fazzi E, Gener B, Haldre M, Lin JP, Livingston JH, Lourenco CM, Marques W Jr, Oades P, Peterson P, Rasmussen M, Roubertie A, Schmidt JL, Shalev SA, Simon R, Spiegel R, Swoboda KJ, Temtamy SA, Vassallo G, Vilain CN, Vogt J, Wermenbol V, Whitehouse WP, Soler D, Olivieri I, Orcesi S, Aglan MS, Zaki MS, Abdel-Salam GM, Vanderver A, Kisand K, Rozenberg F, Lebon P, Crow YJ. Assessment of interferon-related biomarkers in Aicardi-Goutières syndrome associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: a case-control study. Lancet Neurol. 2013a;12:1159 - 69. [PMC free article: PMC4349523] [PubMed: 24183309]
- Rice GI, Kasher PR, Forte GM, Mannion NM, Greenwood SM, Szynkiewicz M, Dickerson JE, Bhaskar SS, Zampini M, Briggs TA, Jenkinson EM, Bacino CA, Battini R, Bertini E, Brogan PA, Brueton LA, Carpanelli M, De Laet C, de Lonlay P, del Toro M, Desguerre I, Fazzi E, Garcia-Cazorla A, Heiberg A, Kawaguchi M, Kumar R, Lin JP, Lourenco CM, Male AM, Marques W Jr, Mignot C, Olivieri I, Orcesi S, Prabhakar P, Rasmussen M, Robinson RA, Rozenberg F, Schmidt JL, Steindl K, Tan TY, van der Merwe WG, Vanderver A, Vassallo G, Wakeling EL, Wassmer E, Whittaker E, Livingston JH, Lebon P, Suzuki T, McLaughlin PJ, Keegan LP, O'Connell MA, Lovell SC, Crow YJ. Mutations in ADAR1 cause Aicardi-Goutières syndrome associated with a type I interferon signature. Nat Genet. 2012;44:1243 - 8. [PMC free article: PMC4154508] [PubMed: 23001123]
- Rice GI, Reijns MA, Coffin SR, Forte GM, Anderson BH, Szynkiewicz M, Gornall H, Gent D, Leitch A, Botella MP, Fazzi E, Gener B, Lagae L, Olivieri I, Orcesi S, Swoboda KJ, Perrino FW, Jackson AP, Crow YJ. Synonymous mutations in RNASEH2A create cryptic splice sites impairing RNase H2 enzyme function in Aicardi-Goutières syndrome. Hum Mutat. 2013b;34:1066 - 70. [PMC free article: PMC3714325] [PubMed: 23592335]
- Rice GI, Rodero MP, Crow YJ. Human disease phenotypes associated with mutations in TREX1. J Clin Immunol. 2015;35:235 - 43. [PubMed: 25731743]
- Richards A, van den Maagdenberg AM, Jen JC, Kavanagh D, Bertram P, Spitzer D, Liszewski MK, Barilla-Labarca ML, Terwindt GM, Kasai Y, McLellan M, Grand MG, Vanmolkot KR, de Vries B, Wan J, Kane MJ, Mamsa H, Schafer R, Stam AH, Haan J, de Jong PT, Storimans CW, van Schooneveld MJ, Oosterhuis JA, Gschwendter A, Dichgans M, Kotschet KE, Hodgkinson S, Hardy TA, Delatycki MB, Hajj-Ali RA, Kothari PH, Nelson SF, Frants RR, Baloh RW, Ferrari MD, Atkinson JP. C-terminal truncations in human 3'-5' DNA exonuclease TREX1 cause autosomal dominant retinal vasculopathy with cerebral leukodystrophy. Nat Genet. 2007;39:1068 - 70. [PubMed: 17660820]
- Samson JF, Barth PG, de Vries JI, Menko FH, Ruitenbeek W, van Oost BA, Jakobs C. Familial mitochondrial encephalopathy with fetal ultrasonographic ventriculomegaly and intracerebral calcifications. Eur J Pediatr. 1994;153:510 - 6. [PubMed: 7957369]
- Sanchis A, Cervero L, Bataller A, Tortajada JL, Huguet J, Crow YJ, Ali M, Higuet LJ, Martinez-Frias ML. Genetic syndromes mimic congenital infections. J Pediatr. 2005;146:701 - 5. [PubMed: 15870678]
- Stephenson JB (2002) Aicardi-Goutieres syndrome - observations of the Glasgow school. Eur J Paediatr Neurol 6 Suppl A:67-70. [PubMed: 12365363]
- Stetson DB, Ko JS, Heidmann T, Medzhitov R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell. 2008;134:587 - 98. [PMC free article: PMC2626626] [PubMed: 18724932]
- Thiele H, du Moulin M, Barczyk K, George C, Schwindt W, Nürnberg G, Frosch M, Kurlemann G, Roth J, Nürnberg P, Rutsch F. Cerebral arterial stenoses and stroke: novel features of Aicardi-Goutières syndrome caused by the Arg164X mutation in SAMHD1 are associated with altered cytokine expression. Hum Mutat. 2010;31:E1836 - 50. [PMC free article: PMC3049152] [PubMed: 20842748]
- Tojo K, Sekijima Y, Suzuki T, Suzuki N, Tomita Y, Yoshida K, Hashimoto T, Ikeda S. Dystonia, mental deterioration, and dyschromatosis symmetrica hereditaria in a family with ADAR1 mutation. Mov Disord. 2006;21:1510 - 3. [PubMed: 16817193]
- Tolmie JL, Shillito P, Hughes-Benzie R, Stephenson JB. The Aicardi-Goutieres syndrome (familial, early onset encephalopathy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis). J Med Genet. 1995;32:881 - 4. [PMC free article: PMC1051740] [PubMed: 8592332]
- Uggetti C, La Piana R, Orcesi S, Egitto MG, Crow YJ, Fazzi E. Aicardi-Goutieres syndrome: neuroradiologic findings and follow-up. AJNR Am J Neuroradiol. 2009;30:1971 - 6. [PMC free article: PMC7051307] [PubMed: 19628626]
- Xin B, Jones S, Puffenberger EG, Hinze C, Bright A, Tan H, Zhou A, Wu G, Vargus-Adams J, Agamanolis D, Wang H. Homozygous mutation in SAMHD1 gene causes cerebral vasculopathy and early onset stroke. Proc Natl Acad Sci U S A. 2011;108:5372 - 7. [PMC free article: PMC3069167] [PubMed: 21402907]
- Yang YG, Lindahl T, Barnes DE. Trex1 exonuclease degrades ssDNA to prevent chronic checkpoint activation and autoimmune disease. Cell. 2007;131:873 - 86. [PubMed: 18045533]
Chapter Notes
Acknowledgments
We would like to thank Dr Gillian Rice for her help in compiling the 基因 variant data.
Author History
Jean Aicardi, MD, FRCP; Hospital Robert-Debré, Paris (2005-2014)
Yanick J Crow, MBBS, BMedSci, MRCP, PhD (2005-present)
John BP Stephenson, DM, FRCP, HonFRCPCH; Royal Hospital for Sick Children, Glasgow (2008-2014)
Revision History
- 22 November 2016 (ma) Comprehensive update posted live
- 13 March 2014 (me) Comprehensive update posted live
- 1 March 2012 (cd) Revision: 靶性性突变分析 for the c.490C>T mutation in TREX1 available clinically
- 19 January 2012 (cd) Revision: deletion/duplication analysis available clinically for SAMHD1, TREX1, RNASEH2A, and RNASEH2B; multigene panels available
- 4 November 2010 (me) Comprehensive update posted live
- 17 April 2008 (me) Comprehensive update posted live
- 29 June 2005 (me) Review posted live
- 1 September 2004 (ja) Original submission