
摘要
临床特征
1p36 缺 失综合征 有典型的颅面部特征,包括平的眉毛、深眼窝、面中部的后缩、鼻嵴凹陷、长人中、尖下颚、晚闭合的前囟(77%)、细发丝(65%)、内眦赘皮(50%),以 及后转、低位和异常的耳朵。其它表现的特征包括短指/先天性指屈曲和短足。100%个体可见不同程度的发育迟缓/智力障碍,95%个体有张力减退。癫痫发 作发生在44%-58% 患者中。其它还包括脑结构异常(88%)、 先天性心脏缺陷(71%)、眼/视力问题(52%)、听力缺损(47%)、骨骼异常(41%)、外生殖器异常(25%)和肾功能异常(22%)。
诊断/实验室检查 根据临床表现可以提示1p36 缺失综合征,而确诊基于1号 (1p36)染色体短臂最远端条带(1p36)的缺失。传统的G显带 细胞遗传学染色体核型分析, FISH,或 (CMA) 染色体芯片(CMA)都可以检测缺失;有些复杂性的缺失只能被CMA检测到。
处理
对症处理:以 康复/教育方案来处理讲话/交流技巧、手语的使用、运动发育、认知和社会技能等方面的问题;婴儿痉挛可使用ACTH;使用常规抗癫痫药物的处理有癫痫表现 的临床表型;特殊的喂养技术和/或设备(包括胃造口)来处理喂养困难;非梗阻性心肌病的标准药物治疗;眼/视力问题、骨骼异常、听力缺损、甲状腺功能减退 和肾功能异常的标准化处理。
监测:随着时间和需求的变化,系统性的随访来调整康复/教育和医疗处理。.
遗传咨询
1p36 缺失综合征是由于几种不同机制引起的1p36区域的缺失造成。大约52%的1p36缺失综合征有1p36染色体末端新发缺失,大约29%有中间段缺失,大 约12%有更复杂的染色体重排(包括一个以上的1p36缺失段,或者缺失和重复同时存在),7%有衍生1号染色体(1p末端着丝粒区域被其它染色体末端替 换而来))。根据缺失的起源机制可以判定家庭成员的风险。已经生育过1q36缺失综合征小孩的家庭,或夫妻双方中有一方已知是1p36相关染色体重排携带 者的,可行产前的检查。
诊断
临床诊断
特殊面容、肌张力低下、精神运动性迟滞和语言能力低下或无语言能力等提示1p36缺失综合征;确诊则依赖1号染色体短臂最远端(1p36)的缺失。
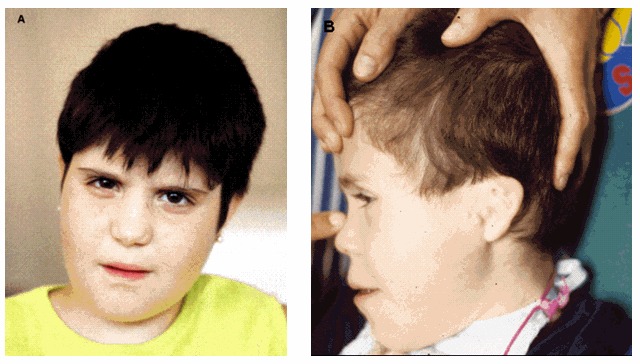
典型面容 作为一个标志性的表型[Battaglia 2005] (see Figure 1),一直以来1p36缺失综合征独特的面部特征都较容易被辨认[Battaglia et al 2008]。面部特征包括平的眉毛、深眼窝、面中部的后缩、鼻嵴凹陷,长人中和尖下颚。其它颅面部特征有小头、短头、内眦赘皮、大(出生时>3cm) 和 晚闭合的前囟,和后转、低位和异常的耳朵[Shapira et al 1997, Heilstedt et al 2003b, Battaglia et al 2008]。
发育迟缓/和智力障碍 在所有个体身上都不同程度的出现。95%会有全身性的肌张力低下[Battaglia et al 2008]。
检测
细胞遗传学分析 有单体性1p36个体已发现的染色体重排分四类,见表 1 [Heilstedt et al 2003b, Gajecka et al 2007]:
注:(1)1p36缺失没有断裂热点,缺失片段大小也不固定。(2)要确定细胞遗传学水平可见的缺失是否是真的末端缺失还是更复杂的重排,要通过专门的分子细胞遗传学技术来完成。(见分子遗传学检测部分)
表1.




分子遗传学检测
导 致1p36缺失综合征唯一已知的原因是在1p36关键区域的基因缺失。『更新:2016年 Fregeau 等发现位于1p36关键区域的RERE基因新发突变的患者表型与1p36缺失患者表型有部分重叠,包括低肌张力、癫痫、行为问题、中枢神经系统结构异常、 眼部异常、先天性心脏病、以及泌尿生殖系统异常。』临床检查
检测策略
确诊先证者诊断的策略: 疑似1p36缺失综合征的合适的检测如下:
注: (1)亚端粒FISH需要根据两个探针信号的存在/缺失来进行判断;因此,FISH(a)不能检测更靠近着丝粒的中间段缺失;(b)不能区分“真正”的末 端缺失和更复杂的重排;或(c)不能明确缺失的范围。而CMA在这三个方面都有潜力。(2)不推荐MLPA(一种缺失/重复的分析的技术)来检测这么大的 缺失。
有1p36缺失风险的妊娠的产前诊断和胚胎植入前遗传学诊断(PGD),需要在确诊了先证者为1p36缺失综合征和/或已知父母是相关平衡易位携带者的前提下进行。
临床特点
临床描述
1p36缺失综合征的主要临床表现相关的频率,见表3。
表3.
1p36缺失综合征的主要临床表现频率
| 临床表现 | 频率 |
|---|---|
| >75% |
| 50%-75% |
| 25%-50% |
| <25% |
智力障碍。 发育迟缓和智力障碍是该综合征的标志性特征。Battaglia et al [2008] 发现25%的个体2岁到7岁开始可以独自行走,宽基步态。约90%个体有严重的智力障碍,而10%的有轻微到中度的认知障碍。75%没有表达性语言,仅限 于几个独立的字,或仅能用一句话的第一个字联系句子其余部分。理解能力仅限于特定的上下文。沟通的试图在早期会有限,但慢慢地会改进,手语能力也会改善。
行为障碍,占50%,包括社交能力差、易怒、自残(咬手和手腕)、刻板行为,和相对少见的多食。
中 枢神经系统障碍,占88%的患者,主要包括侧脑室和蛛网膜下腔的扩张、皮质萎缩、弥漫性脑萎缩;和发育不全,薄,以及完全或部分胼胝体的缺失。其它报道的 异常有髓鞘的形成延迟、白质区多灶性高密度区[Battaglia et al 2008]、脑室周围结节状异位[Neal et al 2006, Descartes et al 2011]和多小脑回[Dobyns et al 2008]。
癫痫 占患者44—58% [Heilstedt et al 2001, Heilstedt et al 2003b, Bahi-Buisson et al 2008, Battaglia et al 2008]。发病年龄从4天到2岁8个月。首次发作是全身性(强直、强直阵挛、阵挛、肌阵挛)或局部发作(简单或复杂性)。几乎20%患有该症的婴儿痉挛 症与脑电图高度节律紊乱有关。婴儿痉挛可能提示癫痫发作类型或者随着其它类型的癫痫发作。标准化药物治疗能控制住大部分类型。然而,在 [Bahi-Buisson et al 2008]的一系列人中,近三分之一出现了耐药性癫痫。
值得注意的是,一些儿童也会发生癫痫窒息[Kanabar et al 2012];有暴发-抑制的早期婴儿癫痫脑病(Ohtahara syndrome,大田原综合征)也在一个个体被报道过[Paciorkowski et al 2011]。
各种的脑电图异常几乎存在在所有患者中[Heilstedt et al 2001, Bahi-Buisson et al 2008, Battaglia et al 2008]。
喂养困难 可能是由于肌张力低下和/或口腔面裂吸允困难,不协调的吞咽及其引起的误吸,和/或胃食管反流和呕吐。研究表明72%的患者会出现轻度到重度口咽的吞咽困难[Heilstedt et al 2003b]。
先天性心脏缺陷 占该病个体的43-71%。报道的结构性心脏缺陷(按频率排列)有房、室间隔缺损、心脏瓣膜异常、动脉导管未闭、法洛四联症、主动脉狭窄、右心室漏斗部狭窄和Ebstein异常[Heilstedt et al 2003b, Battaglia et al 2008]。27%在婴儿期和儿童期有心肌病史。心肌病有23%为非梗阻性型,并在后期会有好转[Battaglia et al 2008]。
眼科异常。 斜视、眼颤、屈光不正和视觉注意力不集中是常见的症状[Heilstedt et al 2003b, Battaglia et al 2008]。偶尔出现白内障、视网膜白化病和视神经缺损畸形[Battaglia et al 2008]。
骨骼异常占40%,包括骨龄延迟、脊柱侧弯、肋骨异常和下肢不对称[Battaglia et al 2008]。
听力缺损, 主要是感觉神经型耳聋,占患者47%-82% [Heilstedt et al 2003b, Battaglia et al 2008]。
泌尿生殖系统畸形 占患者22%,包括单侧肾盂上极积水、异位肾伴右肾囊肿,和单侧肾盂扩张[Battaglia et al 2008]。
少数男性患者中可见尿道下裂、隐睾、和小阴茎[Battaglia et al 2008]。
女性患者中存在小阴唇和阴蒂小,大阴唇肥大和子宫发育不全[Battaglia et al 2008]。
在检测了TSH和T4的水平的患者中,甲状腺功能减退在不同年龄的患者中占15-20%[Heilstedt et al 2003b, Battaglia et al 2008]。
其他。其它被报道过的异常表现:
- 毛细血管扩张性皮肤病变和色素沉着斑[Keppler-Noreuil et al 1995]
- 多指畸形[Keppler-Noreuil et al 1995]
- 先天性椎管狭窄[Reish et al 1995]
- 先天性纤维型不相称性肌病[Okamoto et al 2002]
- 颈部背侧皮冗余[Wang & Chen 2004]
- 肠旋转不良、环状胰和异常排列的胰胆管[Minami et al 2005, Kawashima et al 2011]
- 脂肪肝[Haimi et al 2011]
- 肥厚型幽门狭窄
- 肛门前置或闭锁、钩状或双叶胆囊和小脾脏[Battaglia et al 2008]
- 神经母细胞瘤(3例)[Laureys et al 1990, Biegel et al 1993, Anderson et al 2001]
- 寻常天疱疮(1例)[Halpern et al 2006]
遗传-表型相关性
为了研究1p36缺失综合征的表型变异性,研究人员一直在探索缺失片段大小和临床表现的严重程度之间的相关性。
Wu et al [1999]和Heilstedt et al [2003b]提出该病遗传-表型的完全相关性,确定了某些特征的关键区域,并认为1p36缺失综合征是一个连续性基因缺失综合征。然而Gajecka et al [2007] 在一个大的患者队列中发现缺失片段大小和观察到的临床特征没有相关性,即使是小片段缺失(<3Mb)的患者也会有多数常见症状。Redon et al [2005]提出一个假说,认为1p36缺失综合征的相关特性可能是有位置效应,而不是连续性基因缺失综合征。
发病率
1p36 缺失综合征患病率估计在1/10000到1/5000次分娩,女:男的比例2:1 [Shapira et al 1997, Slavotinek et al 1999, Heilstedt et al 2003a, Battaglia et al 2008]。
遗传相关(等位基因)疾病
除了在GeneReview 中讨论过的表型,没有与1p36关键区拷贝数变异有关的其它已知表型
鉴别诊断
临床表型和1p36缺失综合征的整套面部表型具有该病的代表性。然而,因为和下列疾病有相似的表型,有些个体可能被误诊:
- 瑞特(Rett)综合征是 一种X连锁显性遗传病,其特点有正常出生并在出生后头6-18个月看起来精神运动发育正常,随后在短期内语言能力和运动技能迅速发育倒退。该病的标志是手 的刻板运动和手的失用。自闭症样的特征、突发的恐慌样表现、夜间磨牙症、阵发性呼吸暂停和/或过度换气、步态共济失调和运动不能、震颤、后天的小头畸形也 可发生。疾病会变得相对稳定, 但张力失调和手足畸形随着年龄变得更严重。在Rett综合征女性患者中癫痫发作占50%;全身性强直阵挛性癫痫发作和局部复杂发作最常见。突发不明原因死 亡的发生率增加。核型46,XY的男性可能因严重的新生儿脑病在周岁前死亡。该病的诊断基于典型综合征表现建立的临床诊断标准和/或MECP2的分子检 测。
- 天使(Angelman)综合征(简称AS)主 要特征是严重的发育迟缓/智力障碍、严重的语言障碍、步态共济失调和/或四肢颤抖、以及独特的不合时宜快乐的态度(大笑、微笑和易兴奋)。小头畸形和癫痫 比较常见。诊断由临床特征和分子遗传学检测和/或细胞遗传学分析相结合来进行。已制定了共识的临床诊断标准。在78%的患者的15q11.2-q13染色 体区域内可以检测到父母有别的DNA甲基化印记的异常,病因包括缺失、单亲二倍体和印迹缺陷;少于1%的个体有可见的细胞遗传学重排(易位或倒位)。另外 有11%个体UBE3A基因序列分析可检测出突变。因此,大约90%个体能通过分子遗传学检测(甲基化分析和 UBE3A 基因测序),剩下10%仅有典型的表型特征的个体目前遗传致病机理未知,因而无法完成诊断性检测。
- 普拉德•威利(Prader-Willi)综合征(简称PWS)的 特点是严重的肌无力和婴儿早期的喂养困难,在婴儿后期或儿童早期过度饮食逐步发展成病态的肥胖症(除非在外部控制情况下)。所有患者都有不同程度的认知障 碍、运动和语言能力发育迟缓。特征性的行为表型常见的有暴躁、固执、行为僵化、偷窃、撒谎、操纵他人行为和强迫症。在男性和女性中,都存在性腺功能减退 症,表现为生殖器发育不全、第二性征的发育不完善和多数个体的不孕不育。伴随手小脚小的身材矮小、面部特征性表型、斜视和脊柱侧弯常见。非胰岛素依赖型糖 尿病常在肥胖个体可见。临床诊断标准的共识已制定,但诊断的主要依据是15号染色体Prader-Willi关键区域特殊印记 (PWCR)的DNA的父母有别的甲基化检测。多于99%患者检测出该关键区域仅有母系遗传(不存在父源性区)。甲基化检测对于所有PWS个体的确诊都很 重要,尤其是对表型不典型或年龄太小、临床表型还未能充分表现,难以根据临床表现进行诊断的患者。
- Smith-Magenis综合征(简称SMS)有 独特的面容、发育迟缓、认知障碍和行为异常。面部特征为宽方脸、短头畸形、凸额、一字眉、轻度眼角上翘、眼窝深陷、宽鼻梁、面中部的后缩(以前称为面中部 发育不良)、比较塌陷、短且鼻尖饱满的鼻子、婴儿期小下颌随后随着年龄变为下颌前凸和上唇肉外翻的独特嘴型。SMS具有广泛的认知和适应能力的表型变化, 大多数个体有轻度到中度智力障碍。行为表型有严重的睡眠障碍、刻板性运动、适应环境能力差和自残行为。婴儿期喂养困难、发育停滞、肌张力减退、反射减弱、 长时间的打盹、需唤醒喂食和严重的嗜睡。SMS通过17p11.2中间段缺失或RAI1基因分子遗传检测确诊。常规G显带分析(550条带或更高的分辨 率)能检测到可见的17p11.2中间段缺失。RAI1的突变和缺失是SMS现在已知的唯一原因。FISH或aCGH检测是阴性的个体,可通过测序来检测 该基因。
- Aicardi综合征(简称AIS)的 三联征:胼胝体发育不全、独特的脉络膜视网膜的裂隙和婴儿痉挛症。但是,现在广泛认可的是,另外还有几个重要的表现也在患病的女童身上常见。神经病学检查 可发现小头畸形、躯干张力减退和四肢肌张力痉挛增高。预期中有中度到重度发育迟缓和智力障碍。很多有Aicardi综合征的女孩在三个月内癫痫发作,大多 数一岁前开始癫痫。随后出现有各种临床表现的耐药类型的癫痫。肋椎骨缺陷常见并可在高达1/3的患者中导致脊柱侧弯。其他症状还包括有特征性的特殊面容、 消化困难、手小、血管畸形和皮肤色素异常、肿瘤发生率增加、7到9岁后生长发育迟缓,性早熟或青春期延迟均可见。寿命变化很大,死亡年龄平均值约8.3 岁,死亡年龄中位数约为18.5岁。
管理
初步诊断后的评估
推荐以下评估手段来确立1p36缺失综合征个体的疾病状况和需求:
- 测量生长参数,并在标准生长曲线表上进行标记 注:1p36缺失综合征无特制标准生长曲线表
- 体格检查和神经系统评估
- 认知、语言、运动发育和社交技巧的评估
- 婴儿期心脏检查(听诊、心电图、超声心动图)
- 清醒/睡眠动态多重脑电图检测婴儿痉挛症的高度节律失常
- 评估喂养困难和胃食管反流,并送吞咽问题专家会诊
- 即使无明显异常,在诊断该病后或婴儿期,都请眼科会诊
- 骨骼异常体格检查(脊柱侧弯、下肢不对称);如果有异常,转诊矫形外科和物理治疗评估
- 尽早耳鼻咽喉的综合评价和听力筛查(脑干听觉诱发反应)以适当的干预
- 婴儿期肾功能检查和肾超声检查检测肾结构异常
- 定期的甲状腺功能筛查
- 临床遗传咨询
对症处理
智力障碍。个体化的康复计划中应有运动发育、认知、沟通和社交技能[Battaglia et al 2008]。使用手语提高了沟通技巧,而且并不阻碍语言的发育。早期干预和之后的代之以适当的学校教育是极其关键的。
癫痫发作。高于25%的患者有与脑电图高度节律失常有关的婴儿痉挛症;痉挛对ACTH有反应。
多数患者中,如果及早使用了最合适的药物,所有的癫痫类型都能很好的用标准的抗癫痫药物控制。
喂养困难。对 于这些患者,关注口腔运动技能的喂养疗法是适当的。特殊的喂养技术或设备,比如“Haberman feeder”可用于无腭裂的或未修复腭裂的低肌张力的婴儿。吞咽失调推荐胃导管法。用标准的方式处理胃食管反流。在一个相关研究中,有几个患者做了胃造 口术[Heilstedt et al 2003b]。
先天性心脏缺陷通常不复杂也易于修复。“非阻塞性”心肌病对标准药物治疗(速尿、卡托普利、地高辛)有很好的反应[Battaglia et al 2008]。
眼科异常用标准化方式处理。已报道有64%患者视觉注意力不集中。这些患者可采用适当的康复治疗[Bolognini et al 2005, Battaglia et al 2008]。
骨骼畸形(脊柱侧弯、下肢不对称)需要个体化治疗。建议及早处理(物理治疗和手术)。听力损伤可尝试使用助听器。
其他。结构异常(胃肠道,肾)应以标准的方式处理。甲状腺功能减退也应进行标准化治疗。
监测
系统的随诊使得可以根据患者技能提高或恶化的情况及医疗需求进行康复和治疗方案的调整[Battaglia et al 2008]。
亲属风险的评估
见遗传咨询中的有关风险的评估
还在调研阶段的治疗方法
可通过搜索ClinicalTrials.gov获取各种疾病表型的临床试验的信息。注:该疾病可能没有临床试验。
遗传咨询
遗传咨询的内容是向患者及其家庭提供该病的性质、遗传方式及其可能造成的影响方面的信息,帮助他们做出基于足够背景知识,以及符合个人情况的决定。 接着几个段落是涉及遗传风险评估, 根据家族史和遗传学检测来确定家庭成员的遗传状态。这一段的目的并不是为了解决所有患者可能面临的个人、文化或伦理问题,或者企图替代遗传学专业人员的咨 询工作。-作者ED
遗传方式
遗传或者新发的染色体异常可导致1p36缺失综合征。
家属的风险
先证者的父母
- 父母没有表型但可能是平衡易位的携带者(见表 1)。
- 60%新发的缺失发生在母源性染色体上。
- 大约1/3患者有衍生的1号染色体,该染色体由于平衡易位的亲本染色体分离异常形成。
- 1p36缺失综合征个体的父母应行细胞遗传学分析是否有易位的1p36。不推荐用CMA检测平衡易位。
- 如果先证者的缺失看起来是新发缺失,应对其父母1号染色体进行亚端粒区分析,以判断1号染色体是否存在平衡易位[Heilstedt et al 2003b]。
先证者的同胞
- 同胞的风险取决于父母的遗传状态。
- 若先证者是新发的缺失,同胞和普通人群发病率的风险一致。值得一提的是,生殖细胞嵌合在一个家系被报道[Gajecka et al 2010]。
- 若父母是平衡易位携带者,同胞们1p单体(1p36缺失综合征)或者1p三体相比普通人群发病率的风险值增高。
先证者的后代。未报道过1p36缺失综合征患者生育。
先证者其他家族成员。若父母是染色体重排的携带者,他或她的其他家庭成员也有携带染色体重排的风险。
携带者检测
若先证者的父母有一个是平衡易位的携带者,其他有风险的家庭成员可以用发现他们染色体异常的方法进行检测(染色体分析或亚端粒FISH分析)
遗传咨询的相关问题
具体的咨询内容。1号染色体短臂(1p)和其他染色体易位的具体经验风险值是未知的。
生育规划
- 怀孕前是遗传风险值评估和产前检查可行性分析的最佳时间。
- 给已知有染色体重排、或有这种风险的的年轻成人提供遗传咨询(讨论后代潜在的风险,以及生育的可能方式,比如PGD和产前诊断等)是适当的。
产前检查和胚胎植入前遗传学诊断
高风险的妊娠。在已有1p36缺失综合征患儿,或双亲中有染色体重排的携带者的家庭中,可以考虑对有风险的胎儿进行产前诊断,或考虑(植入前诊断(pre-implantation diagnosis, PGD)。
信息来源
为了该病患者及其家庭成员的方便,GeneReviews的员工已经选择了下述的针对该病的,或者包括其他GeneReviews疾病的患者支持组织和患者注册组织。GeneReviews不为其他组织提供的信息承担责任。选择这些组织的标准,请点击 获取详细信息。
- My46 Trait Profile
- Chromosome Disorder Outreach (CDO)PO Box 724Boca Raton FL 33429-0724Phone: 561-395-4252 (Family Helpline)Email: info@chromodisorder.org
- Unique: The Rare Chromosome Disorder Support GroupG1 The StablesStation Road WestOxted Surrey RH8 9EEUnited KingdomPhone: +44 (0) 1883 723356Email: info@rarechromo.org; rarechromo@aol.com
分子遗传学
在下面的分子生物学表和OMIM表内的信息可能和GeneReview里其他部分的信息不一致:表格里的信息可能更加新-ED
表A。
1p36 缺失综合征:基因和数据库
| 基因 | 染色体位置 | 蛋白 |
|---|---|---|
| 不适用『部分表型可用RERE解释』 | 1p36 | 不适用『部分表型可用RERE解释』 |
数据从以下标准参考资料内整理:基因来自 HGNC;染色体位置、位置名称、关键区域、基因互补群信息来自 OMIM; 蛋白信息来自UniProt. 在此提供了超链接的相关数据库相关的描述,请点击这里:
分子遗传发病机制
没有基因被公认与该综合征的临床表型有关『更新:2016年 Fregeau 等发现RERE基因新发突变的患者表型与1p36缺失患者表型有部分重叠,包括低肌张力、癫痫、行为问题、中枢神经系统结构异常、眼部异常、先天性心脏病、以及泌尿生殖系统异常。』
参考资料
参考文献
- Anderson J, Kempski H, Hill L, Rampling D, Gordon T, Michalski A. Neuroblastoma in monozygotic twins--a case of probable twin-to-twin metastasis. Br J Cancer. 2001;85:493–6. [PMC free article ] [PubMed]
- Bahi-Buisson N, Gutierrez-Delicado E, Soufflet C, Rio M, Cormier Daire V, Lacombe D, Heron D, Verloes A, Zuberi SM, Burglen L, Afenjar A, Moutard LM, Edery P, Dulac O, Nabbout R, Plouin P, Battaglia A. Spectrum of epilepsy in terminal 1p36 deletion syndrome. Epilepsia. 2008;49:509–15. [PubMed]
- Battaglia A. Del 1p36 syndrome: a newly emerging clinical entity. Brain Dev. 2005;27:358–61. [PubMed]
- Battaglia A, Hoyme HE, Dallapiccola B, Zackai E, Hudgins L, McDonald-McGinn D, Bahi-Buisson N, Romano C, Williams CA, Braley LL, Zuberi SM, Carey JC. Further delineation of deletion 1p36 syndrome in 60 patients: a recognizable phenotype and common cause of developmental delay and mental retardation. Pediatrics. 2008;121:404–10. [PubMed]
- Biegel JA, White PS, Marshall HN, Fujimori M, Zackai EH, Scher CD, Brodeur GM, Emanuel BS. Constitutional 1p36 deletion in a child with neuroblastoma. Am J Hum Genet. 1993;52:176–82. [PMC free article ] [PubMed]
- Bolognini N, Rasi F, Coccia M, Ladavas E. Visual search improvement in hemianopic patients after audio-visual stimulation. Brain. 2005;128:2830–42. [PubMed]
- Descartes M, Mikhail FM, Franklin JC, McGrath TM, Bebin M. Monosomy1p36.3 and trisomy 19p13.3 in a child with periventricular nodular heterotopia. Pediatr Neurol. 2011;45:274–8. [PubMed]
- Dobyns WB, Mirzaa G, Christian SL, Petras K, Roseberry J, Clark GD, Curry CJR, McDonald-McGinn D, Medne L, Zackai E, Parsons J, Zand DJ, Hisama FM, Walsh CA, Leventer RJ, Martin CL, Gajecka M, Shaffer LG. Consistent chromosome abnormalities identify novel polymicrogyria loci in 1p36, 2p16.1-p23.1, 4q21.21-q22.1, 6q26-q27, and 21q2. Am J Med Genet A. 2008;146A:1637–54. [PMC free article ] [PubMed]
- Gajecka M, Mackay KL, Shaffer LG. Monosomy 1p36 deletion syndrome. Am J Med Genet C Semin Med Genet. 2007;145C:346–56. [PubMed]
- Gajecka M, Saitta SC, Gentles AJ, Campbell L, Ciprero K, Geiger E, Catherwood A, Rosenfeld JA, Shaikh T, Shaffer LG. Recurrent interstitial 1p36 deletions: evidence for germline mosaicism and complex rearrangement breakpoints. Am J Med Genet A. 2010;152A:3074–83. [PMC free article ] [PubMed]
- Haimi M, Iancu TC, Shaffer LG, Lerner A. Severe lysosomal storage disease of liver in del(1)(p36): a new presentation. Eur J Med Genet. 2011;54:209–13. [PubMed]
- Halpern AV, Bansal A, Heymann WR. Pemphigus vulgaris in a patient with 1p36 deletion syndrome. J Am Acad Dermatol. 2006;55 Suppl 5:98–9. [PubMed]
- Heilstedt HA, Ballif BC, Howard LA, Kashork CD, Shaffer LG. Population data suggest that deletions of 1p36 are a relatively common chromosome abnormality. Clin Genet. 2003a;64:310–6. [PubMed]
- Heilstedt HA, Ballif BC, Howard LA, Lewis RA, Stal S, Kashork CD, Bacino CA, Shapira SK, Shaffer LG. Physical map of 1p36, placement of breakpoints in monosomy 1p36, and clinical characterization of the syndrome. Am J Hum Genet. 2003b;72:1200–12. [PMC free article ] [PubMed]
- Heilstedt HA, Burgess DL, Anderson AE, Chedrawi A, Tharp B, Lee O, Kashork CD, Starkey DE, Wu YQ, Noebels JL, Shaffer LG, Shapira SK. Loss of the potassium channel beta-subunit gene, KCNAB2, is associated with epilepsy in patients with 1p36 deletion syndrome. Epilepsia. 2001;42:1103–11. [PubMed]
- Kanabar G, Boyd S, Schugal A, Bhate S. Multiple causes of apnea in 1p36 deletion syndrome include seizures. Seizure. 2012;21:402–6. [PubMed]
- Kawashima H, Kinjo N, Uejima H, Ioi H, Takekuma K, Nagae I, Ishii K, Itoi T, Numabe H. A case of 1p36 deletion syndrome accompanied with anomalous arrangement of the pancreaticobiliary duct. Pancreas. 2011;40:171–3. [PubMed]
- Keppler-Noreuil KM, Carroll AJ, Finley WH, Rutledge SL. Chromosome 1p terminal deletion: report of new findings and confirmation of two characteristic phenotypes. J Med Genet. 1995;32:619–22. [PMC free article ] [PubMed]
- Laureys G, Speleman F, Opdenakker G, Benoit Y, Leroy J. Constitutional translocation t(1;17)(p36;q12-21) in a patient with neuroblastoma. Genes Chromosomes Cancer. 1990;2:252–4. [PubMed]
- Minami K, Boshi H, Minami T, Tamura A, Yanagawa T, Uemura S, Takifuji K, Kurosawa K, Tsukino R, Izumi G, Yoshikawa N. 1p36 deletion syndrome with intestinal malrotation and annular pancreas. Eur J Pediatr. 2005;164:193–4. [PubMed]
- Neal J, Apse K, Sahin M, Walsh CA, Sheen VL. Deletion of chromosome 1p36 is associated with periventricular nodular heterotopia. Am J Med Genet A. 2006;140:1692–5. [PubMed]
- Okamoto N, Toribe Y, Nakajima T, Okinaga T, Kurosawa K, Nonaka I, Shimokawa O, Matsumoto N. A girl with 1p36 deletion syndrome and congenital fiber type disproportion myopathy. J Hum Genet. 2002;47:556–9. [PubMed]
- Paciorkowski AR, Thio LL, Rosenfeld JA, Gajecka M, Gurnett CA, Kulkarni S, Chung WK, Marsh ED, Gentile M, Reggin JD, Wheless JW, Balasubramian S, Kumar R, Christian SL, Marini C, Guerrini R, Maltsev N, Shaffer LG, Dobyns WB. Copy number variants and infantile spasms: evidence for abnormalities in ventral forebrain development and pathways of synaptic function. Eur J Hum Genet. 2011;19:1238–45. [PMC free article ] [PubMed]
- Redon R, Rio M, Gregory SG, Cooper RA, Fiegler H, Sanlaville D, Banerjee R, Scott C, Carr P, Langford C, Cormier-Daire V, Munnich A, Carter NP, Colleaux L. Tiling path resolution mapping of constitutional 1p36 deletions by array-CGH: contiguous gene deletion or "deletion with positional effect" syndrome? J Med Genet. 2005;42:166–71. [PMC free article ] [PubMed]
- Reish O, Berry SA, Hirsch B. Partial monosomy of chromosome 1p36.3: characterization of the critical region and delineation of a syndrome. Am J Med Genet. 1995;59:467–75. [PubMed]
- Shapira SK, McCaskill C, Northrup H, Spikes AS, Elder FF, Sutton VR, Korenberg JR, Greenberg F, Shaffer LG. Chromosome 1p36 deletions: the clinical phenotype and molecular characterization of a common newly delineated syndrome. Am J Hum Genet. 1997;61:642–50. [PMC free article ] [PubMed]
- Slavotinek A, Shaffer LG, Shapira SK. Monosomy 1p36. J Med Genet. 1999;36:657–63. [PMC free article ] [PubMed]
- Wang BT, Chen M. Redundant skin over the nape in a girl with monosomy 1p36 caused by a de-novo satellited derivative chromosome: a possible new feature? Clin Dysmorphol. 2004;13:107–9. [PubMed]
- Wu YQ, Heilstedt HA, Bedell JA, May KM, Starkey DE, McPherson JD, Shapira SK, Shaffer LG. Molecular refinement of the 1p36 deletion syndrome reveals size diversity and a preponderance of maternally derived deletions. Hum Mol Genet. 1999;8:313–21. [PubMed]
本章节的注解
作者历史
Agatino Battaglia, MD (2008-今)
Lisa G Shaffer, PhD, FACMG; Signature Genomic Laboratories (2008-2013)
更新历史
- 2013/6/6 (Lisa G Shaffer ) 系统性更新发布到公开网页上
- 2008/2/1 (Lisa G Shaffer ) 内容发布到公开网页上
- 2007/8/24 (Agatino Battaglia) 最早稿件