
概述
临床特征。
EZH2 相关过度生长包括 EZH2 相关Weaver 综合征,即 EZH2和高身材。尽管大多数被诊断患有杂合的 EZH2 致病性变异的个体是由于临床怀疑而被鉴定为 Weaver 综合征,少数通过分子遗传学检测在先证者的家庭成员或过度生长的个体但无韦弗综合征的临床表现鉴定出致病性变异。因此,与杂合 EZH2 致病性变异相关的表型谱的范围尚不清楚。
Weaver综合征的特点是身材高大、智力不等(从正常智力到严重智力残疾)、特征性面部外观以及一系列相关的临床特征,包括骨龄高、协调性差、皮肤柔软、手指弯曲和/或脚趾,脐疝,音调异常,婴儿期低声嘶哑。脑部 MRI 已发现一些与 EZH2 相关过度生长个体存在异常。神经母细胞瘤在具有 EZH2 杂合的致病性变异的个体中的发生频率略有增加,但数据不足以确定绝对风险。目前没有证据表明其他恶性肿瘤(包括血液系统恶性肿瘤)的发生频率增加。
诊断/测试。
EZH2相关过度生长的诊断是分子遗传学检测检测到杂合的 胚系EZH2 致病性变异。
管理。
表现的治疗:对于发育迟缓和/或学习障碍的个体,可能需要转介学习/行为/言语评估和支持。有时,脚趾弯曲可能需要手术释放。物理治疗可能对那些因韧带松弛或关节挛缩而出现关节疼痛的人有益。脊柱侧弯的治疗是常规治疗。应针对其他临床问题进行适当的专家转诊。
监测:定期对患有 EZH2 相关Weaver综合征的幼儿进行医疗随访,以监测发育进展、弯曲指(用于解决/改善)和/或肌张力减退;对没有医疗并发症的年龄较大的儿童/青少年进行医疗随访的频率可能会降低。如果存在脊柱侧弯,请按照骨科医生的建议进行监测。虽然目前的数据不支持特定的肿瘤监测计划,但临床医生应该对任何可能与肿瘤(特别是神经母细胞瘤)相关的发现进行调查。
怀孕管理:家庭及其医疗保健提供者应该意识到受累的婴儿可能很大,以便制定适当的分娩计划。
遗传咨询。
EZH2 相关过度生长以常染色体显性遗传方式遗传;然而,许多de novo 胚系致病性 EZH2 变异出现。具有 EZH2 致病性变异个体的每个孩子都有 50% 的机会遗传致病性变异;无法预测遗传 EZH2 致病性变异的个体中表型的严重程度。如果在 受累的家庭成员中鉴定出致病性变异,则可能进行 产前诊断,用于妊娠风险增加和 植入前遗传诊断。
GeneReview 谱
| EZH2 相关过度生长:包括表型 |
|---|
|
有关同义词和过时的名称,请参见 Nomenclature.
- 1.
对于其他遗传原因 表型 见 Differential Diagnosis.
诊断
提示性发现
在具有以下临床和影像学表现的个体中应怀疑 EZH2 相关过度生长 [Tatton-Brown et al 2013]:
临床发现
- 身材高大(身高≥+2 SD)
- 大头畸形(头围≥+2 SD)
- 智力残疾
- 特征面容
- 三岁以下的儿童:下颌后缩、大而多肉的耳朵、下巴“卡住”外观并伴有水平皮肤皱褶,有时还有中央酒窝
- 宽阔的额头
- 眼睛间距大
- 杏仁状睑裂
注意:特征性的面部外观(在年轻时最明显)随着时间的推移而演变; 因此,回顾儿童时期的照片可能有助于临床医生做出临床诊断。 - 协调性差
- 柔软而有弹性的皮肤
- 手指和/或脚趾弯曲(见注解)
- 偶尔严重到需要手术复位的脐疝
- 异常张力(中枢性张力减退和/或外周张力亢进)(见注解)
- 嘶哑、低沉的哭声(有时被描述为安静的哭声)
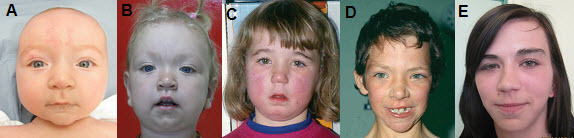
Figure 1.
患有 EZH2 相关 Weaver 综合征的年幼儿童出现的下颌后缩通常会随着年龄的增长而消退。 在所有年龄段的人中,睑裂通常呈杏仁状,眼睛间距很大。
注意:可能需要详细的病史以确定这些发现是否存在于新生儿期/婴儿期,因为它们可以在整个童年时期解决/改善。
影像学发现。平片上的骨龄晚期
脑部核磁共振。在许多人中,尚未进行脑部 MRI,因此无法获得成像数据。在 10 人中发现的一项或多项异常包括: 孤立的脑室扩大(5 人);脑室扩大和脑室周围白质软化 (1);脑室周围白质软化症 (1);小脑梗塞 (1);和小脑发育不全和神经元迁移缺陷(多小脑回)伴或不伴脑回 (2) [Al-Salem et al 2013, Tatton-Brown et al 2013].
建立诊断
EZH2相关过度生长的诊断是通过分子遗传学检测在先证者中鉴定出杂合的 胚系EZH2致病性变异(见Table 1)。
分子基因检测方法可以进行单基因检测、multigene panel检测、全基因组的检测(外显子组测序, exome array, 基因组测序 ):
- 单基因测试。单基因检测要求临床医生识别Weaver综合征/EZH2相关过度生长表型 ,要求同时检测分子遗传学检测EZH2。然而,鉴于表型广泛且面部特征微妙,即使对于经验丰富的畸形学家来说,这也可能具有挑战性。EZH2 的序列分析检测小的基因内缺失/插入和 错义, nonsense和剪接位点变异;通常,不会检测到外显子或全基因删除/重复。如果序列分析未鉴定出致病性变异,则可以考虑进行靶向deletion/duplication analysis以检测基因内缺失或重复。注意:据报道,在几个可能由于 Weaver 综合征导致生长过度的个体中存在全基因或部分基因缺失 [Imagawa et al 2017, Suri & Dixit 2017].
- 多基因套餐测试。更常见的是,患有 EZH2 相关过度生长的个体在使用多基因套餐进行测试后被诊断为与智力障碍相关生长(身高和/或头围)过度(见Differential Diagnosis)。 EZH2 经常包含在此类多基因套餐中。这种套餐可能以最合理的成本鉴定疾病的遗传原因,同时减少意义不确定 变异和不能解释表型的变异出现。
注:(1)panel 中包含的基因和实验室用于每个基因的检测敏感性可能随时间而变化。 (2) 一些多基因 panel 可能包含与本 GeneReview 中讨论的疾病无关的基因。 (3) 在一些实验室,panel 选项可能包括定制的实验室设计的panel 和/或定制的以表型为重点的 外显子组分析,其中包括临床医生指定的基因。 (4) panel 中使用的方法可能包括序列分析, deletion/duplication analysis和/或其他非基于序列的测试。
有关多基因套餐的介绍,请单击here。有关订购基因检测的临床医生的更多详细信息,请参见here。
Table 1
用于 EZH2 相关过度生长的分子遗传学检测
| 基因 1 | 方法 | 此法测出致病变异的比例 2 |
|---|---|---|
| EZH2 | 测序 3 | 迄今为止报告的大多数变异 4 |
| 基因靶向deletion/duplication analysis 5 | 见注解 6 |
- 1.
染色体 位点和蛋白质信息见 Table A. Genes and Database 。
- 2.
有关在该基因中检测到的等位基因变异的信息见 Molecular Genetics 。
- 3.
- 4.
- 5.
基因靶向deletion/duplication analysis检测基因内缺失或重复。 使用的方法可以包括: quantitative PCR,长程PCR和多重连接依赖性探针扩增(MLPA)和设计用于检测单 外显子缺失或重复的基因靶向。
- 6.
两个人有完全或部分包含 EZH2 的缺失 [Imagawa et al 2017, Suri & Dixit 2017].
临床特征
临床表现
胚系EZH2 致病变异相关的表型谱很广,谱系中有典型的 Weaver 综合征和高身材。尽管多数被诊断为杂合的EZH2 致病性变异的个体是由于 Weaver 综合征而被临床鉴定的,少数通过分子遗传学检测 先征者的家族成员或有过度生长但无Weaver 综合征表现的个体而诊断 [Tatton-Brown et al 2011, Gibson et al 2012]。因此,杂合 EZH2 致病变异的表型谱的范围尚不清楚。虽然数据仍然有限,但迄今为止报告的 54 名 Weaver 综合征患者的临床关联总结如下 [Tatton-Brown et al 2011, Gibson et al 2012, Al-Salem et al 2013, Tatton-Brown et al 2013, Usemann et al 2016, Suri & Dixit 2017, Lui et al 2018]。分母反映了可获得数据的个人数量。
生长。根据 23 名新生儿的可用数据,平均出生身长比平均值高 2.2 个标准差(+2.2 SD),范围为 -0.5 SD 至 +4.9 SD; 45 名新生儿的平均出生体重为 +1.7 SD,范围为 -1.6 SD 至 +4.6 SD [Tatton-Brown et al 2013].
高身材是一个几乎一致的发现:47/52 人的身高至少比平均值高两个标准差(年龄 1-70 岁)。值得注意的是,四个身高低于 +2 SD 的人中有 3 个在幼儿时就已经很高了。平均出生后身高为+3.5 SD。
在可获得信息的 45 人中,24 人的头围小于 +2 SD,21 人患有大头畸形,头围高达 +5.5 SD。
认知特征。 50 人可获得有关认知功能的信息。八人智力正常; 42 人患有不等程度智力障碍 (ID),其中包括:
- 轻度 ID (24/50)。孩子们在普通学校上学,需要一些额外的帮助——例如,教育需求声明——但他们应该像成年人一样独立生活,并且很可能有自己的家庭。
- 中等 ID (14/50)。儿童发展语言并在普通教育中需要高水平的支持,但更有可能进入有特殊教育需要的人的学校。虽然不太可能像成年人一样独立生活,但他们可能会住在庇护所或有一些额外的支持。
- 严重 ID (2/50)。个人在上学期间需要特殊教育,并且可能在成年后需要大量支持。
- 未分类的 ID (2/50)。提供的信息不足以做出决定。
行为问题。 有传闻报道了包括自闭症谱系障碍、恐惧症和焦虑在内的行为问题。
神经病学。在 6 名个体中报告的脑室扩大通常与正常的 CSF 压力相关,不需要分流[Tatton-Brown et al 2013]. 其他脑部 MRI 发现包括神经元沟回缺陷(肿块/多小脑回;2 人)、脑室周围白质软化(2 人)和小脑异常(2 人)。
脑部 MRI 异常者的智力障碍是:
- 6 例轻度(脑室扩大 [4]、脑室周围白质软化 [1] 和小脑发育不全 [1]);
- 3例中度 (脑室周围白质软化伴脑室扩大 [1] 和 孤立的脑室扩大 [2]);
- 1例重度 ,有多小脑回症和厚脑回症;相比之下, Al-Salem et al [2013]报道的患有多小脑回的个体具有正常的发育和身体不对称(左侧小于右侧),左侧反射活跃且肌力增加。
- 注:一名脑室扩大患者的智力残疾程度未报告。
四个人有高热惊厥。
骨骼特征
- 骨龄提前。在评估的 29 人中,所有人的骨龄均已超前。
- 9 人报告了脊柱侧弯,3 人报告了胸部异常(漏斗或鸡胸)。脊柱侧弯的范围从严重(儿童早期发病需要手术干预)到轻度(需要监测但没有治疗干预)。
- 弯曲指。一些受累的人手指弯曲,一些脚趾弯曲,还有一些手指和脚趾弯曲。有时脚趾弯曲需要手术矫正。
- 马蹄足。六个人有马蹄内翻足,从两个人的固定和双侧(需要手术)到三个人的轻度(单侧,通过物理治疗解决)
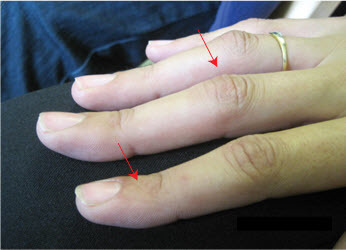
Figure 2.
患有杂合 EZH2 致病性变异的 22 岁女性远端指间关节轻度过度伸展和近端指间关节屈曲
结缔组织
- 韧带松弛。虽然与关节过度活动和扁平足相关的韧带松弛很常见,但除非合并关节疼痛,否则通常不会报告。经常报告有 EZH2 相关过度生长的个体协调性差,这可能(至少部分)归因于韧带松弛。
- 皮肤19/37受累的孩子的皮肤摸起来柔软而有弹性。
- 脐疝,见于 21/44 的儿童,8 名大到需要在 新生儿期进行手术。
肌张力异常。一般而言,如果存在异常张力(张力减退、张力亢进或混合中枢张力减退和外周张力亢进),儿童期就会消退。
- 22/45 人报告肌张力减退(主要是中枢性)。
- 13/41 报告了肌张力亢进(主要是外周表现为肢体僵硬,反射活跃)。
注意:据报道,有 5 名患有外周性张力亢进的个体也患有中枢性张力减退。
喂养不良。据报道,10/28 的新生儿喂养不良,其中一名需要鼻胃管喂养两周。尽管喂养不良可能是由于新生儿肌张力减退,但这仅在三名喂养不良的婴儿中报道。
嘶哑、低沉的哭声。 10/27受累的婴儿出现嘶哑、低沉的哭声。
肿瘤。据报道,54 名受累的患者中有 4 人患有肿瘤[Tatton-Brown et al 2013, Usemann et al 2016].
- 一名患有 c.2233G>A 致病性变异的男孩在 13 岁时患上了前 T 细胞非霍奇金淋巴瘤。在 25 岁时,他仍然很好,没有复发或其他肿瘤。
- 一名患有 c.2044G>A 致病性变异的男孩在 13 个月大时被诊断出患有急性淋巴细胞白血病和神经母细胞瘤,两者均对治疗有反应;他七岁了。
- 一名患有 c.458A>G 致病性变异的女孩在 4 岁时被诊断出患有神经母细胞瘤。
- 一名患有 c.395C>T 致病性变异的女孩在 16 岁时被诊断出患有急性髓性白血病和继发性噬血细胞性淋巴组织细胞增多症。
其他临床特征 在少数个体中报告的其他临床特征(因此,可能与 EZH2 致病性变异无关)包括在内以保证完整性:
- 牛奶咖啡斑(2 人)、血管瘤(4 人)
- 远视(远视)(3)、近视(1)、斜视(3)
- 隐睾 (1)、鞘膜积液 (2)、尿道下裂 (1)
- 腭裂 (3)
- 听力损失 (3) – 传导性和感音神经性
- 心脏异常 (4),包括二尖瓣脱垂 (1)、室间隔缺损 (2) 和动脉导管未闭 (1)
- 胃食管反流 (1)、裂孔疝 (1)
- 新生儿低血糖症 (2)
- 新生儿低钙血症 (1)
基因型-表型相关性
因为在具有杂合的截断致病变异或杂合错义致病变异的个体中观察到整个表型谱的发现,在保守的 SET 结构域之内或之外(参见Molecular Genetics,正常基因产物),在少数EZH2 相关过度生长的个体中没有genotype-phenotype correlations 。
外显率
目前数据不足以确定EZH2 胚系致病变异的外显率。然而,鉴于某些具有 EZH2 致病性变异的人的表型的复杂性,一些 EZH2 致病性变异的外显率可能会降低 [Tatton-Brown et al 2013].
命名法
Weaver 综合征以 David Weaver 的名字命名,他报告了两个男孩骨成熟加速、面容异常和弯曲指 [Weaver et al 1974].
尽管 NSD1 的致病性变异(Sotos syndrome的原因)曾被报道会导致 Weaver 综合征 [Douglas et al 2003], 但这种关联已被驳斥 [Tatton-Brown et al 2005].
患病率
由于 EZH2 致病性变异最近才被证明会导致 Weaver 综合征,而具有轻度表型的个体可能会在临床诊断时漏诊,因此目前很难估计 Weaver 综合征的患病率。
遗传相关(等位基因)疾病
除本 GeneReview 中讨论的表型外,没有已知表型与 EZH2 中的胚系致病变异相关。
在没有任何其他 Weaver 综合征发现的情况下发生的散发性肿瘤(包括造血系统恶性肿瘤)通常在 EZH2 中含有 胚系中不存在的体细胞变异。 在这些情况下,这些肿瘤的易感性是不可遗传的。 有关更多信息,请参阅Cancer and Benign Tumors.
诊断
Weaver综合征的鉴别诊断中要考虑的条件总结 Table 2.
Table 2
Weaver综合征鉴别诊断中需要考虑的疾病
| 疾病 | 基因 / 分子机理 | MOI | 临床特征 | |
|---|---|---|---|---|
| 相同点 | 鉴别点 | |||
| Sotos syndrome | NSD1 1 | AD 2 |
| Sotos 综合征的面部特征:
|
| Malan syndrome (OMIM 614753) | NFIX | AD |
| Malan综合征:
|
| DNMT3A 过度生长(Tatton-Brown-Rahman) syndrome (OMIM 615879) | DNMT3A4 | AD |
| 在 DNMT3A 过度生长综合征中:
|
| Beckwith-Wiedeman syndrome (BWS) | 在11p15.5 5印记的2个异常调节转录基因 | 脚注 6 |
| 在 BWS 中:
|
| Simpson-Golabi-Behmel syndrome type 1 (SGBS1) | GPC3 | XL |
| SGBS1:
|
| Marfan syndrome | FBN1 | AD |
| Marfan 综合征:
|
| Congenital contractural arachnodactyly (CCA; Beals syndrome) | FBN2 | AD |
| CCA:
|
- 1.
NSD1 致病基因( Sotos syndrome病因) 曾经被认为是Weaver syndrome病因 [Douglas et al 2003]. 但是, 这种观点后来被推翻 [Tatton-Brown et al 2005].
- 2.
- 3.
- 4.
- 5.
BWS 的临床怀疑可以通过测试来证实,该测试显示 11p15 生长调节区域的正常印记失调 [Choufani et al 2010]:印记中心 2 的甲基化丢失(IC2;参见 Beckwith-Wiedeman Syndrome, Figure 1了解详细的分子机制)关于母体等位基因(约 50% 受累的个体); 11p15 的单亲二体性(约 20% 的受影响个体); 印记中心 1 甲基化(IC12;参见 Beckwith-Wiedeman Syndrome, Figure 1了解详细的分子机制)(在约 5% 的受影响个体中); 或 CDKN1C 母体拷贝中的致病变异(在 BWS 的散发性病例中占 5%-10%;在家族性病例中≤40%)。 在大约 20% 的临床诊断为 BWS 的个体中,潜在的分子异常尚未阐明。
- 6.
大约 85% 的 BWS 患者没有 BWS 家族史; 大约 15% 的人有与父母一致的家族史,以常染色体显性遗传 方式遗传。
- 7.
处理
以下信息代表美国个人的典型评估和管理建议; 标准建议可能因国家/地区而异[作者,个人观察]。
初步诊断后的评估
为了确定被诊断患有杂合的EZH2 致病性变异的个体的疾病程度和需求,建议进行Table 3中总结的评估(如果未作为导致诊断的评估的一部分进行)。
Table 3
ZH2 相关过度生长个体初步诊断后的推荐评估
| 系统/关注 | 评估 | 备注 |
|---|---|---|
神经病学 | 发育评估 | 包括运动、言语/语言评估、一般认知和职业技能 |
| 肌张力 | 低张力和混合性低/高张力常见 | |
| 如果怀疑癫痫发作:脑 MRIEEG
| |
| 精神病学/行为学 | 评估 ASD 和其他行为问题 | |
| 一般体检 | 测量身高、体重、头围 | |
| 肌肉骨骼 | ||
| 泌尿生殖系统 | 评估隐睾、鞘膜积液、尿道下裂 | |
| 心血管 | 心脏听诊 | 用于结构性心脏异常证据的基线超声心动图 |
| 恶性肿瘤 | 评估潜在的恶性肿瘤,尤其是神经母细胞瘤和血液系统恶性肿瘤 | 虽然不建议进行具体监测,但建议对任何可能的肿瘤相关症状进行早期调查。 |
ASD = 自闭症谱系障碍
治疗表现
Table 4
EZH2相关过度生长个体表现的治疗
| 表现/关注 | 治疗 | 注意事项/其他 |
|---|---|---|
| 发育迟缓和/或学习障碍 | 教育支持 | 可能会指示学习/行为/演讲评估和支持的推荐。 |
| 弯曲指 | 手术治疗 | 有时脚趾弯曲可能需要手术干预。. |
| 理疗 | 可能是有益的 | |
| 肌张力异常 | 理疗 | 可能是有益的 |
| 韧带松弛 | 理疗 | 可减轻继发于韧带松弛的关节疼痛 |
| 脊柱侧弯 | 进一步评估和监测 | 转诊至骨科医生 |
如果通过病史和/或检查发现其他临床问题,则应进行适当的专家转诊。
全球发育障碍/智力障碍管理问题
年龄 0-3 岁。建议转诊至早期干预计划以获得职业、身体、言语和喂养治疗。在美国,早期干预是一项联邦资助的计划,在所有州都可以使用。
年龄3-5岁。在美国,建议通过当地公立学区进行发展学前教育。在安置之前,会进行评估以确定所需的服务和治疗,并制定个性化教育计划 (IEP)。
年龄 5-21 岁
- 在美国,应由当地公立学区制定基于个人功能水平的 IEP。受影响的儿童可以留在公立学校系统直到 21 岁。
- 应从 12 岁开始讨论过渡计划,包括财务、职业/就业(如果可行)和医疗安排。发育儿科医生可以为过渡到成年期提供帮助。
所有年龄段。建议咨询发育儿科医生,以确保适当的社区、州和教育机构的参与,并支持父母最大限度地提高生活质量。
建议根据受累的个人的需要考虑采用私人支持疗法。关于治疗类型的具体建议可以由发育儿科医生提出。
在美国:
- 建议注册发育障碍管理局 (DDA)。 DDA 是一个为合格个人提供服务和支持的公共机构。资格因州而异,但通常由诊断和/或相关的认知/适应性障碍决定。
- 收入和资源有限的家庭也可能有资格为其残疾子女获得补充保障收入 (SSI)。
运动功能障碍
粗大运动功能障碍
- 建议进行物理治疗以最大限度地提高活动能力。
- 根据需要考虑使用耐用的医疗设备(例如,轮椅、助行器、浴椅、矫形器、自适应婴儿车)。
精细运动功能障碍。对于影响适应功能的精细运动技能困难,如喂养、梳洗、穿衣和写作,建议进行职业治疗。
口腔运动功能障碍。假设个体可以安全地通过口进食,对于因口腔运动控制不佳而难以进食的受累的个体,建议采用喂食疗法(通常由职业治疗师或语言治疗师提供)。
沟通问题。考虑为有表达语言困难的个人评估替代交流方式(例如, Augmentative and Alternative Communication [AAC])。
监测Table 5
对 EZH2 相关过度生长个体的推荐监测
| 系统/问题 | 评估 | 频率 1 |
|---|---|---|
| 肌肉骨骼 | 儿科医生的监测,以解决/改善弯曲指和/或肌张力减退症 | 定期随访,频率取决于严重程度 |
| 如果存在脊柱侧弯,按照骨科建议进行监测 | ||
| 恶性肿瘤 | 神经母细胞瘤监测:目前的建议包括对可能与肿瘤相关的任何症状保持临床警惕和彻底调查。 2 注意:神经母细胞瘤监测一直不一致,没有数据支持监测方式、筛查间隔或持续时间。 | |
| 各种各样的/ 其他 | 监测发展进度和教育需求 | 定期跟进,频率取决于严重程度 |
临床遗传学评估 | 在诊断时,不久后回答进一步的问题,并在适当的时候支持生殖决定(即,让父母讨论再发风险或让受累的个人讨论后代风险) |
- 1.
对于没有医疗并发症的年龄较大的儿童/青少年,临床医生可能希望检查频率低于年龄较大的儿童。
- 2.
目前的数据表明,具有杂合的 胚系EZH2 致病变异的个体发生神经母细胞瘤的相对风险略有增加。 尽管数字太小,无法量化绝对肿瘤风险,但似乎很低(见Clinical Description,肿瘤)
评估有风险的亲属
有关为遗传咨询目的而对高危亲属进行测试的相关问题,请参阅Genetic Counseling。
孕期管理
一般来说,母亲和/或胎儿患有杂合的EZH2 致病性变异的妊娠并不复杂。 家庭及其医疗保健提供者应该意识到受累的婴儿可能很大,以便制定适当的分娩计划; 此外,应提供有关 EZH2 相关过度生长表型的信息。
正在研究的疗法
在美国和EU Clinical Trials Register中搜索 ClinicalTrials.gov,以获取有关各种疾病和病症的临床研究信息。 注意:可能没有针对这种疾病的临床试验。
遗传咨询
遗传咨询是向个人和家庭提供有关遗传疾病的性质、遗传和影响的信息的过程,以帮助他们做出明智的医疗和个人决定。 以下部分涉及遗传风险评估以及使用家族史和基因检测来阐明家庭成员的遗传状况。 本节并非旨在解决个人可能面临的所有个人、文化、道德问题或替代咨询遗传学专业人士。 —编者。
遗传模式
EZH2相关过度生长以常染色体显性遗传方式遗传
家族成员风险
先证者的父母
- 一些被诊断患有 EZH2 相关过度生长的人有受累的父母。
- 一些被诊断为与 EZH2 相关的过度生长的个体由于新发 EZH2 致病性变异而患有这种疾病。 由 de novo致病变异引起的 EZH2 相关过度生长的比例是未知的。 在被鉴定为 EZH2 致病性变异的 48 名个体的系列中 [Tatton-Brown et al 2013]:
- 14例家族性病例.
先证者的同胞。先证者同胞的风险取决于先证者父母的遗传状况。
- 如果测试显示先证者的父母有 EZH2 致病性变异,则同胞遗传该变异的风险为 50%。无法预测遗传 EZH2 致病性变异的个体的表型。
- 如果在任一亲本的白细胞 DNA 中都无法检测到 EZH2 致病性变异,则由于理论上存在亲本胚系嵌合的可能性,因此估计同胞的 再发风险为 1% [Rahbari et al 2016].
如果父母未进行 EZH2 致病性变异检测且临床上不受影响,则先证者同胞的风险尚不清楚,因为父母 外显率降低(或胚系嵌合)的可能性.
先证者的后代。患有 EZH2 相关过度生长的个体的每个孩子都有 50% 的机会继承 EZH2致病性变异 。
其他家庭成员。对其他家庭成员的风险取决于 先证者父母的状态:如果父母有 EZH2致病性变异,他或她的家庭成员可能处于危险之中。
相关遗传咨询问题
具有明显de novo 致病性变异的家族的注意事项。当具有常染色体显性遗传病的先证者的父母都没有在先证者或疾病的临床证据中发现的致病性变异时,致病性变异很可能是新发的。然而,也可以探索非医学解释,包括非生物学父亲或生育(例如,辅助生殖)和未公开的收养。
家庭计划
DNA 银行是存储 DNA(通常从白细胞中提取)以备将来使用。因为将来测试方法和我们对基因、等位基因变异和疾病的理解可能会有所改善,所以应该考虑储存受累的个体的 DNA。
产前检测和胚胎植入前遗传学诊断
一旦在 受累的高危妊娠和家庭成员的产前检测中鉴定出 EZH2 致病性变异,就有可能进行植入前遗传诊断。
医疗专业人员和家庭内部对于产前检测的使用可能存在观点差异,特别是如果考虑进行检测是为了终止妊娠而不是早期诊断。 虽然大多数中心会认为有关产前检查的决定是父母的选择,但讨论这些问题是适当的。
资源
GeneReviews 工作人员选择了以下特定疾病和/或支持组织和/或登记处,以造福患有这种疾病的个人及其家人。 GeneReviews 不对其他组织提供的信息负责。 有关选择标准的信息,请单击 here.
- National Library of Medicine Genetics Home Reference
- Child Growth Foundation21 Malvern DriveWest Midlands B76 1PZUnited KingdomPhone: 020 8995 0257Email: nfo@childgrowthfoundation.org
- MAGIC Foundation4200 Cantera Drive #106Warrenville IL 60555Phone: 800-362-4423 (Toll-free Parent Help Line); 630-836-8200Fax: 630-836-8181Email: contactus@magicfoundation.org
分子遗传
Molecular Genetics 和 OMIM 表格中的信息可能与 GeneReview 中其他地方的信息不同:表格可能包含更新的信息。 —编者。
Table A
EZH2 相关过度生长:基因和数据库
| 基因 | 染色体定位 | 蛋白质 | 数据库 | HGMD | ClinVar |
|---|---|---|---|---|---|
| EZH2 | 7q36 | Histone-lysine N-methyltransferase EZH2 | EZH2 database | EZH2 | EZH2 |
Table B
EZH2 相关过度生长的 OMIM 条目 (View All in OMIM)
基因结构。有多种转录改变。最长的 NM_004456.4包含 20 个外显子;第一个 外显子是非编码的。有关基因和蛋白质信息的详细摘要,请参见Table A,基因。
致病性变异。在解释序列变异时要考虑的因素:
Table 6
GeneReview 中讨论的 EZH2 致病性变异
| DNA 核酸改变 | 蛋白质改变 | 测序 |
|---|---|---|
| c.395C>T | p.Pro132Leu | NM_004456 NP_004447 |
| c.458A>G | p.Tyr153Cys | |
| c.2044G>A | p.Ala682Thr | |
| c.2233G>A | p.Glu745Lys |
关于变异分类的注意事项:表中列出的变异由作者提供。 GeneReviews工作人员尚未独立验证变异的分类。
关于命名法的注释:GeneReviews遵循人类基因组变异学会(varnomen- .hgvs.org )的标准命名惯例。 有关术语的解释,请参见Quick Reference。
正常基因产物。 EZH2(NP_004447.2), 由最长的转录本编码,是一个 751 个氨基酸的 histone甲基转移酶,具有一个关键的 SET (su(var)3-9, zeste增强子, trithorax) 结构域,一个前SET CXC 结构域,和两个额外的 SANT(Sw13、Ada2、N-cor TFIIIB)域 [Wu et al 2013]. 与 SUZ12 EED 结合(形成多梳阻遏复合物 2 [PCR2]),EZH2 通过组蛋白 3 的赖氨酸残基 27 的 甲基化来抑制转录,该功能由 SET 结构域催化 [Cao et al 2002].
异常的基因产物。 EZH2 错义和截短变异导致 Weaver 综合征的蛋白质改变和机制目前尚不清楚。然而,值得注意的是,致病性错义变异是主要的突变机制。迄今为止发现的少数致病性截断变体靶向最终的外显子,因此很可能逃避无意义介导的 RNA 衰变。目前的证据表明,Weaver 综合征可能是由组蛋白甲基转移酶功能受损引起的 [Cohen et al 2016, Lui et al 2018]。
癌症和良性肿瘤
体细胞激活和失活单和双等位基因的EZH2 致病变异已在各种造血系统恶性肿瘤中被发现。特别值得注意的是,已经在淋巴瘤,特别是大 B 细胞淋巴瘤和滤泡性淋巴瘤中发现了一种针对 Tyr646 残基的复发性激活致病性变异。在预后不良的骨髓增生异常和骨髓增殖性肿瘤中发现了整个基因的失活致病变异[Morin et al 2010, Sneeringer et al 2010, Chase & Cross 2011].
尽管体细胞失活EZH2 致病性变异与发病较晚的慢性骨髓增生/发育异常综合征之间存在关联,但在 EZH2 相关过度生长的个体中,这些恶性肿瘤的发生频率似乎并未增加。这一发现的可能解释:
- 骨髓增殖性/发育异常综合征的年龄大于目前鉴定为胚系(构成性)EZH2致病性变异的大多数个体的年龄。
- EZH2体细胞致病变异导致恶性肿瘤和EZH2 胚系致病变异导致过度生长的机制不同。
- 迄今为止,已鉴定出相对较少的具有 EZH2 胚系致病变异的个体。
References
Literature Cited
- Al-Salem A, Alshammari MJ, Hassan H, Alazami AM, Alkuraya FS. Weaver syndrome and defective cortical development: a rare association. Am J Med Genet A. 2013;161A:225 - 7. [PubMed: 23239504]
- Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039 - 43. [PubMed: 12351676]
- Chase A, Cross NC. Aberrations of EZH2 in cancer. Clin Cancer Res. 2011;17:2613 - 8. [PubMed: 21367748]
- Choufani S, Shuman C, Weksberg R. Beckwith-Wiedemann syndrome. Am J Med Genet C Semin Med Genet. 2010;154C:343 - 54. [PubMed: 20803657]
- Cohen AS, Yap DB, Lewis ME, Chijiwa C, Ramos-Arroyo MA, Tkachenko N, Milano V, Fradin M, McKinnon ML, Townsend KN, Xu J, Van Allen MI, Ross CJ, Dobyns WB, Weaver DD, Gibson WT. Weaver syndrome-associated EZH2 protein variants show impaired histone methyltransferase function in vitro. Hum Mutat. 2016;37:301 - 7. [PMC free article: PMC4832389] [PubMed: 26694085]
- Cottereau E, Mortemousque I, Moizard MP, Bürglen L, Lacombe D, Gilbert-Dussardier B, Sigaudy S, Boute O, David A, Faivre L, Amiel J, Robertson R, Viana Ramos F, Bieth E, Odent S, Demeer B, Mathieu M, Gaillard D, Van Maldergem L, Baujat G, Maystadt I, Héron D, Verloes A, Philip N, Cormier-Daire V, Frouté MF, Pinson L, Blanchet P, Sarda P, Willems M, Jacquinet A, Ratbi I, Van Den Ende J, Lackmy-Port Lis M, Goldenberg A, Bonneau D, Rossignol S, Toutain A. Phenotypic spectrum of Simpson-Golabi-Behmel syndrome in a series of 42 cases with a mutation in GPC3 and review of the literature. Am J Med Genet C Semin Med Genet. 2013;163C:92 - 105. [PubMed: 23606591]
- Douglas J, Hanks S, Temple IK, Davies S, Murray A, Upadhyaya M, Tomkins S, Hughes HE, Cole TR, Rahman N. NSD1 mutations are the major cause of Sotos syndrome and occur in some cases of Weaver syndrome but are rare in other overgrowth phenotypes. Am J Hum Genet. 2003;72:132 - 43. [PMC free article: PMC378618] [PubMed: 12464997]
- Gibson WT, Hood RL, Zhan SH, Bulman DE, Fejes AP, Moore R, Mungall AJ, Eydoux P, Babul-Hirji R, An J, Marra MA., FORGE Canada Consortium. Chitayat D, Boycott KM, Weaver DD, Jones SJ. Mutations in EZH2 cause Weaver syndrome. Am J Hum Genet. 2012;90:110 - 8. [PMC free article: PMC3257956] [PubMed: 22177091]
- Golabi M, Rosen L. A new X-linked mental retardation-overgrowth syndrome. Am J Med Genet. 1984;17:345 - 58. [PubMed: 6538755]
- Imagawa E, Higashimoto K, Sakai Y, Numakura C, Okamoto N, Matsunaga S, Ryo A, Sato Y, Sanefuji M, Ihara K, Takada Y, Nishimura G, Saitsu H, Mizuguchi T, Miyatake S, Nakashima M, Miyake N, Soejima H, Matsumoto N. Mutations in genes encoding polycomb repressive complex 2 subunits cause Weaver syndrome. Hum Mutat. 2017;38:637 - 48. [PubMed: 28229514]
- Klaassens M, Morrogh D, Rosser EM, Jaffer F, Vreeburg M, Bok LA, Segboer T, van Belzen M, Quinlivan RM, Kumar A, Hurst JA, Scott RH. Malan syndrome: Sotos-like overgrowth with de novo NFIX sequence variants and deletions in six new patients and a review of the literature. Eur J Hum Genet. 2015;23:610 - 5. [PMC free article: PMC4402637] [PubMed: 25118028]
- Lui JC, Barnes KM, Dong L, Yue S, Graber E, Rapaport R, Dauber A, Nilsson O, Baron J. Ezh2 mutations found in the Weaver overgrowth syndrome cause a partial loss of H3K27 histone methyltransferase activity. J Clin Endocrinol Metab. 2018;103:1470 - 8. [PMC free article: PMC6276576] [PubMed: 29244146]
- Malan V, Rajan D, Thomas S, Shaw AC, Louis Dit Picard H, Layet V, Till M, van Haeringen A, Mortier G, Nampoothiri S, Puseljic S, Legeai-Mallet L, Carter NP, Vekemans M, Munnich A, Hennekam RC, Colleaux L, Cormier-Daire V. Distinct effects of allelic NFIX mutations on nonsense-mediated mRNA decay engender either a Sotos-like or a Marshall-Smith syndrome. Am J Hum Genet. 2010;87:189 - 98. [PMC free article: PMC2917711] [PubMed: 20673863]
- Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, Paul JE, Boyle M, Woolcock BW, Kuchenbauer F, Yap D, Humphries RK, Griffith OL, Shah S, Zhu H, Kimbara M, Shashkin P, Charlot JF, Tcherpakov M, Corbett R, Tam A, Varhol R, Smailus D, Moksa M, Zhao Y, Delaney A, Qian H, Birol I, Schein J, Moore R, Holt R, Horsman DE, Connors JM, Jones S, Aparicio S, Hirst M, Gascoyne RD, Marra MA. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010;42:181 - 5. [PMC free article: PMC2850970] [PubMed: 20081860]
- Priolo M, Schanze D, Tatton-Brown K, Mulder PA, Tenorio J, Kooblall K, Acero IH, Alkuraya FS, Arias P, Bernardini L, Bijlsma EK, Cole T, Coubes C, Dapia I, Davies S, Di Donato N, Elcioglu NH, Fahrner JA, Foster A, González NG, Huber I, Iascone M, Kaiser AS, Kamath A, Liebelt J, Lynch SA, Maas SM, Mammì C, Mathijssen IB, McKee S, Menke LA, Mirzaa GM, Montgomery T, Neubauer D, Neumann TE, Pintomalli L, Pisanti MA, Plomp AS, Price S, Salter C, Santos-Simarro F, Sarda P, Segovia M, Shaw-Smith C, Smithson S, Suri M, Valdez RM, Van Haeringen A, Van Hagen JM, Zollino M, Lapunzina P, Thakker RV, Zenker M, Hennekam RC. Further delineation of Malan syndrome. Hum Mutat. 2018;39:1226 - 37. [PMC free article: PMC6175110] [PubMed: 29897170]
- Rahbari R, Wuster A, Lindsay SJ, Hardwick RJ, Alexandrov LB, Turki SA, Dominiczak A, Morris A, Porteous D, Smith B, Stratton MR. UK10K Consortium, Hurles ME. Timing, rates and spectra of human germline mutation. Nat Genet. 2016;48:126 - 33. [PMC free article: PMC4731925] [PubMed: 26656846]
- Schanze D, Neubauer D, Cormier-Daire V, Delrue MA, Dieux-Coeslier A, Hasegawa T, Holmberg EE, Koenig R, Krueger G, Schanze I, Seemanova E, Shaw AC, Vogt J, Volleth M, Reis A, Meinecke P, Hennekam RC, Zenker M. Deletions in the 3' part of the NFIX gene including a recurrent Alu-mediated deletion of exon 6 and 7 account for previously unexplained cases of Marshall-Smith syndrome. Hum Mutat. 2014;35:1092 - 100. [PubMed: 24924640]
- Sneeringer CJ, Scott MP, Kuntz KW, Knutson SK, Pollock RM, Richon VM, Copeland RA. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc Natl Acad Sci U S A. 2010;107:20980 - 5. [PMC free article: PMC3000297] [PubMed: 21078963]
- Suri T, Dixit A. The phenotype of EZH2 haploinsufficiency-1.2-Mb deletion at 7q36.1 in a child with tall stature and intellectual disability. Am J Med Genet A. 2017;173:2731 - 5. [PubMed: 28696078]
- Tatton-Brown K, Douglas J, Coleman K, Baujat G, Cole TR, Das S, Horn D, Hughes HE, Temple IK, Faravelli F, Waggoner D, Turkmen S, Cormier-Daire V, Irrthum A, Rahman N. Genotype-phenotype associations in Sotos syndrome: an analysis of 266 individuals with NSD1 aberrations. Am J Hum Genet. 2005;77:193 - 204. [PMC free article: PMC1224542] [PubMed: 15942875]
- Tatton-Brown K, Hanks S, Ruark E, Zachariou A, Duarte Sdel V, Ramsay E, Snape K, Murray A, Perdeaux ER, Seal S, Loveday C, Banka S, Clericuzio C, Flinter F, Magee A, McConnell V, Patton M, Raith W, Rankin J, Splitt M, Strenger V, Taylor C, Wheeler P, Temple KI, Cole T, Douglas J, Rahman N. Germline mutations in the oncogene EZH2 cause Weaver syndrome and increased human height. Oncotarget. 2011;2:1127 - 33. [PMC free article: PMC3282071] [PubMed: 22190405]
- Tatton-Brown K, Murray A, Hanks S, Douglas J, Armstrong R, Banka S, Bird LM, Clericuzio CL, Cormier-Daire V, Cushing T, Flinter F, Jacquemont ML, Joss S, Kinning E, Lynch SA, Magee A, McConnell V, Medeira A, Ozono K, Patton M, Rankin J, Shears D, Simon M, Splitt M, Strenger V, Stuurman K, Taylor C, Titheradge H, Van Maldergem L, Temple IK, Cole T, Seal S., Childhood Overgrowth Consortium. Rahman N. Weaver syndrome and EZH2 mutations: clarifying the clinical phenotype. Am J Med Genet A. 2013;161A:2972 - 80. [PubMed: 24214728]
- Tatton-Brown K, Seal S, Ruark E, Harmer J, Ramsay E, Del Vecchio Duarte S, Zachariou A, Hanks S, O'Brien E, Aksglaede L, Baralle D, Dabir T, Gener B, Goudie D, Homfray T, Kumar A, Pilz DT, Selicorni A, Temple IK, Van Maldergem L, Yachelevich N., Childhood Overgrowth Consortium. van Montfort R, Rahman N. Mutations in the DNA methyltransferase gene DNMT3A cause an overgrowth syndrome with intellectual disability. Nat Genet. 2014;46:385 - 8. [PMC free article: PMC3981653] [PubMed: 24614070]
- Tatton-Brown K, Zachariou A, Loveday C, Renwick A, Mahamdallie S, Aksglaede L, Baralle D, Barge-Schaapveld D, Blyth M, Bouma M, Breckpot J, Crabb B, Dabir T, Cormier-Daire V, Fauth C, Fisher R, Gener B, Goudie D, Homfray T, Hunter M, Jorgensen A, Kant SG, Kirally-Borri C, Koolen D, Kumar A, Labilloy A, Lees M, Marcelis C, Mercer C, Mignot C, Miller K, Neas K, Newbury-Ecob R, Pilz DT, Posmyk R, Prada C, Ramsey K, Randolph LM, Selicorni A, Shears D, Suri M, Temple IK, Turnpenny P, Val Maldergem L, Varghese V, Veenstra-Knol HE, Yachelevich N, Yates L, Rahman N, et al. The Tatton-Brown-Rahman syndrome: a clinical study of 55 individuals with de novo constitutive DNMT3A variants. Wellcome Open Res. 2018;3:46. [PMC free article: PMC5964628] [PubMed: 29900417]
- Usemann J, Ernst T, Schäfer V, Lehmberg K, Seeger K. EZH2 mutation in an adolescent with Weaver syndrome developing acute myeloid leukemia and secondary hemophagocytic lymphohistiocytosis. Am J Med Genet A. 2016;170A:1274 - 7. [PubMed: 26762561]
- Weaver DD, Graham CB, Thomas IT, Smith DW. A new overgrowth syndrome with accelerated skeletal maturation, unusual facies, and camptodactyly. J Pediatr. 1974;84:547 - 52. [PubMed: 4366187]
- Wu H, Zeng H, Dong A, Li F, He H, Senisterra G, Seitova A, Duan S, Brown PJ, Vedadi M, Arrowsmith CH, Schapira M. Structure of the catalytic domain of EZH2 reveals conformational plasticity in cofactor and substrate binding sites and explains oncogenic mutations. PLoS One. 2013;8:e83737. [PMC free article: PMC3868588] [PubMed: 24367611]
Chapter Notes
Revision History
- 2 August 2018 (bp) Comprehensive update posted live
- 6 August 2015 (me) Comprehensive update posted live
- 18 July 2013 (me) Review posted live
- 23 January 2013 (ktb) Original submission