
概述
临床特征。
Lynch(林奇)综合征是由错配碱基修复基因的胚系致病性变异引起的,并与肿瘤的微卫星不稳定性(MSI)相关,其特征是结肠癌、子宫内膜、卵巢癌、胃癌、小肠癌、肝胆道癌、泌尿道癌、脑和皮肤肿瘤的风险增加。林奇综合征病人一生中患癌的风险分别为:结直肠癌52%-82% (中位诊断年龄 44-61岁);女性子宫内膜癌25%-60% (中位诊断年龄48-62岁);胃癌 6%-13% (中位诊断年龄56岁);卵巢癌4%-12%(中位诊断年龄42.5 岁;大约30%在40岁之前发病)。其他林奇综合征相关癌症的发病风险相对较低,但是仍然显著高于普通人群。
诊断/检测方法。
如果一个病人的家族史符合Amsterdam(阿姆斯特丹)标准,而且肿瘤具有微卫星不稳定性 (MSI);或者一个个体或者家族的分子遗传学检测找到了错配碱基修复基因(MMR基因包括MLH1、MSH2、MSH6、PMS2)的胚系致病性变异就可以诊断为林奇综合征。MLH1和MSH2的胚系致病性变异大约占林奇综合征家庭所有致病性变异的90%; MSH6 的致病性变异大约占7%-10%;PMS2的致病性变异的比例小于5%。EPCAM基因的胚系缺失 (并不是一个错配修复基因) 可以使MSH2失活,大约占林奇综合征患者的1%。
林奇综合征的基因检测最好是分步进行:
治疗。
对症治疗: 对于结肠癌,推荐行全结肠切除术+回肠直肠吻合术。
主要症状的预防“: 对于已经明确诊断为林奇综合征的患者,在没有出现结肠癌之前,不推荐行预防性的结肠切除术,因为常规的结肠镜检查就是一种有效的预防措施。在完成生育之后,可以考虑行预防性的子宫和卵巢切除术(在发展为癌症之前)。
监测:建议从20-25岁或者家族中最早的癌症诊断年龄提前5年(取两者中更早的时间点)开始,每1-2年行结肠镜检查,并且切除还未癌变的息肉。子宫内膜癌、卵巢癌、胃癌、十二指肠癌和泌尿系统癌症的监测效能还不清楚。
需避免的药物/环境: 吸烟。
风险亲属评估: 不推荐对小于18岁的亲属进行林奇综合征的基因检测;但是,因为推荐在家族中最早的患癌年龄之前10年开始进行筛查,分子遗传学检测和结肠镜筛查在某些家庭中可能需要在18岁之前进行。
遗传咨询。
林奇综合征遵循常染色体显性遗传模式。大多数林奇综合征患者是从父母那里遗传来的这个疾病。但是,由于外显率不全、癌症的发病年龄不一、筛查和预防性手术导致癌症风险下降,并不是所有的具有MMR基因致病性变异的林奇综合征患者都具有父母患癌症史。林奇综合征患者的任何一个小孩都有50%的可能性会遗传这个致病性变异。如果家族的致病性变异已知,就可以进行产前诊断。虽然林奇综合征有很多治疗方法,但是那些典型林奇综合征患者前来申请做产前诊断的并不多。
诊断
如果一个病人的家族史符合阿姆斯特丹(Amsterdam)标准,而且肿瘤具有 微卫星 不稳定性 (MSI);或者一个个体或者家族的分子遗传学检测找到了错配碱基修复基因(MMR,包括MLH1, MSH2, MSH6和PMS2)或者EPCAM基因的胚系 致病性变异 ,就可以诊断为林奇综合征。
在1990遗传性非息肉病性结直肠癌国际协作组建立了第一个临床标准,Amsterdam标准,用于定义遗传性非息肉病性结直肠癌(HNPCC,林奇综合征),以便识别和研究林奇综合征家庭。这些标准后面又做了一些修改 (Amsterdam II 标准),将其它的林奇综合征相关的肿瘤都包含进来了 (Table 1).
表1. HNPCC临床诊断的Amsterdam 标准和Amsterdam II 标准
| Amsterdam标准 1 | Amsterdam II 标准2 |
|---|---|
|
|
- 1.
- 2.
- 3.
父母、子女或兄弟姐妹
- 4.
结直肠癌、子宫内膜癌、胃癌、小肠癌、肝胆系统肿瘤、肾盂癌或输尿管癌
尽管Amsterdam标准是MMR基因的 胚系 致病性变异的一个重要预测指标,但是却漏诊了很多具有MMR基因的 胚系 致病性变异的林奇综合征患者。因此,不能依赖家族史和Amsterdam标准来识别MMR基因的 胚系 致病性变异。
注: 任何个体,只要找到了四个MMR基因的其中一个基因的胚系 致病性变异,就可以确诊为林奇综合征,不管他是否具有家族史。
检测
肿瘤检测
可以对肿瘤组织进行微卫星不稳定性(MSI)和/或免疫组织化学(IHC)检测 ,以便:
- 确定Lynch综合征的可能性;
- 确定林奇综合征患者最可能出现哪种基因的胚系致病性变异。
Table 2总结了用于解释肿瘤检测结果的可能原因和基于肿瘤检测结果推荐的额外检测。
组织类型
- 取结直肠癌组织进行检测显然是最好的,但是如果没有癌组织,可以考虑对腺瘤性息肉进行检测。然而,在林奇综合征相关息肉中出现异常MSI或IHC的概率更低。来自69个具有致病性变异个体的109个息肉的研究发现,仅有79%的腺瘤表现出MMR蛋白的表达丧失。高度不典型增生的息肉与致病性变异状态的一致性明显高于低度不典型增生的息肉[Walsh et al 2012]。
- 该检测也可用于筛查有MMR缺陷的子宫内膜癌 [Backes et al 2009]。
肿瘤组织的免疫组织化学检测(IHC)。IHC检测错配碱基修复基因表达的蛋白质产物的存在与否。 MMR基因产物以二聚体的形式发挥作用:MSH2蛋白可与MSH6或MSH3蛋白复合,MLH1蛋白与PMS2或PMS1蛋白复合。 MSH6和PMS2蛋白质在不与复合物配对时是不稳定的。因此,MSH2中的胚系致病性变异通常导致蛋白质MSH2和 MSH6的同时性表达缺失,而MLH1中的胚系致病性变异将导致蛋白质MLH1 和PMS2的同时性表达缺失。然而,MSH6和PMS2的胚系致病性变异通常不会导致MSH2或MLH1表达缺失,因为这些蛋白质仍然存在于其他配对中[Bellizzi & Frankel 2009].
IHC检测的优点:
- IHC检测可有效检测MMR蛋白表达缺失的肿瘤。 目前已经证实,MLH1、MSH2、MSH6和PMS2的抗体,在具有胚系致病性变异个体的肿瘤中的敏感性为92%[Shia 2008].
- IHC检测在大多数中心都很容易实施,并且技术上易于执行。
- IHC检测在大多数个体中有助于识别MMR基因,其中最有可能发现胚系致病性变异或沉默基因表达的体细胞改变[de Leeuw et al 2000, Cunningham et al 2001],从而显著减少分子遗传学检测的成本。
IHC测试的局限性:
- 组织固定和其他技术问题的变化可能导致染色弱或模棱两可的染色[Shia 2008]。
- 一些错义胚系变异可能不会导致蛋白质表达产物的缺失[Wahlberg et al 2002, Bellizzi & Frankel 2009]。
- 在小组织样本上进行可能不太可靠 [Zhang 2008]。
肿瘤组织的微卫星不稳定性(MSI)检测。错配碱基修复(MMR)途径中的基因负责识别和修复细胞生长和分裂时发生的单核苷酸错配和环状的插入或缺失环 [Gruber & Kohlmann 2003]。与错配修复有关的基因缺陷导致细胞中致病性体系突变的累积,这可能导致细胞发生恶变。
微卫星是具有重复核苷酸序列(例如,AAAAA或CGCGCGCG)的DNA片段,当错配碱基修复基因功能受损时,它们特别容易获得错误。与正常组织相比,具有错配修复基因功能缺陷的细胞发生的癌症表现出微卫星核苷酸重复数的不一致,这一发现被称为“微卫星不稳定性”。见Figure 1.
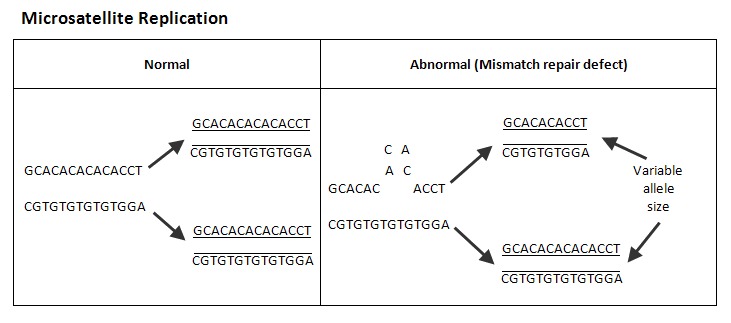
通常是通过比较肿瘤组织和正常组织中的一组微卫星标记来评估MSI。1997年NCI共识会议建议测试五个标记的核心组:BAT25、BAT26、D2S123、D5S346和D17S250 [Boland et al 1998]。这5个微卫星标记仍然是最常用的,它包括两个单核苷酸和三个双核苷酸重复的标记。然而,许多实验室现在正在使用各种其它的标记 [Hegde et al 2014]。根据MSI检测的结果,将肿瘤分类如下 [Boland et al 1998, Hegde et al 2014]:
- 高度微卫星不稳定(MSI-H):核心组五个标记中的两个或两个以上显示不稳定性,或其他标记物组中超过30%的标记物显示不稳定。
- 低度微卫星不稳定(MSI-L):核心组五个标记中,只有一个显示不稳定性,或其他标记物组中少于30%的标记物显示不稳定。
- 微卫星稳定(MSS):核心组或其他标记物组中,0个(或0%)标记物显示不稳定。
注意:尽管一些临床实验室在进行MSI检测时使用了额外的标记,但是除了 Boland et al [1998]设计的标记之外,对标记缺乏共识。
- 结肠肿瘤。当没有足够的癌组织用于MSI检测时,也可用腺瘤行MSI检测。Lynch综合征相关腺瘤的研究表明,与侵袭性癌症相比,腺瘤发生MSI的概率略低,约80%的腺瘤为MSI-H(见 Table 3)。具有高度不典型增生的腺瘤比早期息肉更可能表现出MSI [Iino et al 2000]。
- 子宫内膜癌。大约20%-30%的子宫内膜癌表现出MSI,并且与结肠癌一样,大多数是体细胞MLH1启动子甲基化的结果[Hampel et al 2006]。
MSI检测的优点:
- MSI检测是确定哪些肿瘤由MMR缺陷引起的有效方法。研究表明,肿瘤MSI检测用于筛查胚胎MMR基因变异个体的敏感性为93%[Shia 2008]。
- 当IHC研究给出假阴性结果时(例如,因为检测中不包括合适的抗体;蛋白质是存在的,但是没有功能),MSI检测可能是阳性的(即MSI-H,鉴定肿瘤是由MMR缺陷引起的)[Shia 2008]。
- MSI检测需要非常少的组织[Zhang 2008]。
- MSI检测具有高度可重复性[Zhang 2008]。
MSI检测的局限性:
- 它可能并非在所有中心都可用,因为它需要显微切割和分子分析 [Bellizzi & Frankel 2009]。
- 在一些肿瘤中,由于一些技术的问题,比如极端粘液性肿瘤中缺乏DNA,无法检测到MSI[Hampel et al 2005]。
- Lynch综合征相关肿瘤的一小部分不会出现MMR缺陷的证据[Shia 2008]。
- 它不会降低分子检测的成本,因为它无助于识别最可能发生突变的基因 。
- 结肠肿瘤。 BRAF的致病性变异,最常见的是NM_004333.4: c.1799T> A(p.Val600Glu,或V600E),发生在15%的结直肠癌中。 BRAF的致病性变异在Lynch综合征相关癌症中被认为是罕见的。因此,一般而言,BRAF致病性变异的存在排除了Lynch综合征的诊断 [Bellizzi & Frankel 2009, Bouzourene et al 2010]。
- 子宫内膜癌。 BRAF的致病性变异在散发性子宫内膜癌中并不常见。因此,BRAF检测无助于区分散发性的子宫内膜癌与Lynch综合征相关的子宫内膜癌 [Kawaguchi et al 2009]。
肿瘤组织的甲基化分析。大部分MSI是由于MLH1启动子区的体细胞甲基化引起的,它可以沉默肿瘤组织中的基因表达。因此,MLH1启动子甲基化的发现常常有助于排除Lynch综合征的诊断。
然而,甲基化对剩余的功能等位基因的失活也可能是导致基因纯合失活的“第二次打击”,导致具有胚系MLH1致病性变异的个体发生肿瘤[Bellizzi & Frankel 2009, Niessen et al 2009]。因此,在早发性结肠癌、强烈家族史或其他危险因素的个体中,MLH1启动子超甲基化的存在不能排除Lynch综合征的诊断。
已发现功能性MSH2等位基因的体细胞甲基化是约24%的MSH2相关癌症中的第二次打击。然而,尚未发现MSH2的甲基化是散发性结直肠癌的原因 [Nagasaka et al 2010]。
表2. 肿瘤组织检测结果的解读和进一步检测一览表
| 肿瘤组织检测 1 | 可能的病因 | 进一步的检测 2 | ||||||
|---|---|---|---|---|---|---|---|---|
| 免疫组化 (IHC) | MSI | BRAF V600E 3 | MLH1 启动子甲基化 | |||||
| MLH1 | MSH2 | MSH6 | PMS2 | |||||
| + | + | + | + | MSS/ MSI-L | 散发性癌 | 无 4 | ||
| + | + | + | + | MSI-H | 任一MMR基因的胚系突变 | 考虑MLH1和MSH2的胚系基因突变检测,然后再检测MSH6和PMS2。 | ||
| MSI-H | 散发性癌或者任一MMR基因的 胚系突变 | 考虑行免疫组化导向的基因胚系突变检测。 如果没有行IHC,则先行MLH1和MSH2的胚系基因突变检测,然后再检测MSH6和PMS2。 | ||||||
| – | + | + | – | 散发性癌 MLH1基因的胚系突变 | 考虑行 BRAF基因突变检测3/或者MLH1的启动子甲基化检测; 如果没有检测到BRAF基因的致病性变异或者MLH1的启动子甲基化,或者没有行BRAF/甲基化检测,则行MLH1的胚系突变检测。 | |||
| – | + | + | – | 阳性 | 散发性癌 | 无 4 | ||
| – | + | + | – | 阴性 | 阳性 | 散发性癌 罕见, MLH1基因的胚系突变或者MLH1的组成性表观突变。 | 无,如果发病年龄很年轻或者有强烈的家族史,则考虑行MLH1的胚系突变检测;如果发病年龄很年轻,还应考虑行MLH1的组成性表观突变检测。 | |
| – | + | + | – | 阴性 | 阴性 | MLH1基因的胚系突变 | MLH1基因的胚系突变检测 | |
| + | – | – | + | MSH2基因的胚系突变 EPCAM基因的胚系突变 罕见, 胚系基因的胚系突变 | MSH2基因的胚系突变检测 如果没有检测到MSH2的致病性变异,则考虑行EPCAM基因突变检测 (如果还没有做的话)。 如果没有检测到MSH2或EPCAM的致病性变异(包含大片段缺失和重组的检测),则考虑行MSH6基因的胚系突变检测。 | |||
| – | + | + | + | MLH1基因的胚系突变 | MLH1基因的胚系突变检测 | |||
| + | + | + | – | PMS2基因的胚系突变 MLH1基因的胚系突变 | PMS2基因的胚系突变检测 如果没有检测到PMS2的致病性变异,则行MLH1基因的胚系突变检测 | |||
| + | – | + | + | MSH2基因的胚系突变 | MSH2基因的胚系突变检测 | |||
| + | + | – | + | MSH6基因的胚系突变 MSH2基因的胚系突变 | MSH6基因的胚系突变检测 如果没有检测到MSH6基因的致病性变异,则行MSH2基因的胚系突变检测。 | |||
NCCN [2013] (点击 here 获取全文; 需要注册)
空单元格表示未进行测试或结果可能不会影响测试策略。
MSI = 微卫星不稳定
MSS = 微卫星稳定
- +
= 蛋白质染色正常
– = 蛋白质染色缺失
Neg = 阴性
Pos = 阳性
- 1.
肿瘤测试策略适用于结直肠癌和子宫内膜癌。关于肿瘤测试在其他Lynch综合征肿瘤中功效的数据有限。
- 2.
有关胚系致病性变异测试的信息,请参阅Molecular Genetic Testing和Table 3。
- 3.
测试不适用于结直肠癌以外的肿瘤。
- 4.
分子遗传学检测
基因。已经鉴定了五种基因(MLH1、MSH2、MSH6、PMS2和EPCAM [以前称为TACSTD1]),他们的胚系致病性变异体破坏了错配碱基修复(MMR)途径并引起Lynch综合征(参见Table 3)。
注意:虽然EPCAM不是MMR基因,但它的3'端的胚系大片段缺失将导致相邻的下游MSH2通过高甲基化沉默[Niessen et al 2009, Goel et al 2011, Kuiper et al 2011]。
附加位点异质性的证据。在一些Lynch综合征家庭中有一些胚系MLH3、MSH3、EXO1、PMS1或TGFBR2变异的病例报告;然而,Lynch综合征中这些基因中等位基因变异体的临床意义尚未确定[Lu et al 1998, Peltomäki 2003]。虽然这些基因和其他基因可能在 错配碱基修复过程中发挥作用,但作为Lynch综合征评估的一部分,这些基因的突变检测仍然没有确定的作用 [Thompson et al 2004, Ou et al 2009, Duraturo et al 2011]。可以在www.omim.org上找到关于这些基因在Lynch综合征中可能发挥作用的证据的简洁讨论:
在怀疑患有Lynch综合征的个体中测试这些基因中胚系变异体的临床效用是未知的。
临床检测
表3. 用于Lynch综合征的胚系分子遗传学检测的小结
| 基因 1 | Lynch综合征归因于该基因的致病性变异的比例 | 检测方法 |
|---|---|---|
| MLH1 | 50% 2, 3 | 序列分析 4 |
| 大片段缺失/重复分析 5, 6 | ||
| MSH2 | 40% 2 | 序列分析 4 |
| 大片段缺失/重复分析 5, 6 | ||
| MSH6 | 7%-10% 7 | 序列分析 4 |
| 大片段缺失/重复分析 5, 6 | ||
| PMS2 | <5% 8 | 序列分析 4, 10 |
| 大片段缺失/重复分析 5, 10 | ||
| EPCAM (TACSTD1) | ~1%-3% 9 | 序列分析 4 |
| 大片段缺失/重复分析 5, 6, 11 |
- 1.
参见Table A. Genes and Databases 中的有关染色体 位点和蛋白质。有关等位基因变体的信息,请参阅Molecular Genetics。
- 2.
- 3.
- 4.
- 5.
- 6.
大片段的缺失和遗传重排占MSH2中致病性变异体的20%,MLH1中致病性变异体的5%,PMS2中致病性变异体的20%,MSH6中致病性变异体的7%,以及EPCAM中致病性变异体的100% [Wijnen et al 1998, Charbonnier et al 2000, Wagner et al 2003, Plaschke et al 2004, Senter et al 2008]。
- 7.
- 8.
- 9.
- 10.
由于PMS2和假基因之间的高度同源性,因此难以对该基因的突变进行测试和解释。当在一个家庭中怀疑PMS2致病性变异时,应选择遵循PMS2分析ACMG指南且具有测试该基因专业知识的实验室进行检测 [Hegde et al 2014]。
- 11.
EPCAM的胚系缺失导致通过高甲基化沉默相邻的MSH2等位基因。相邻的MSH2等位基因本身未发生突变(参见Molecular Genetic Pathogenesis)。
测试的特征。有关测试特征(包括敏感性和特异性)的信息,请参见 Clinical Utility Gene Card [Rahner et al 2013]。
测试的策略
证实/确定先证者中Lynch综合征的诊断。要查看由国家综合癌症网络(NCCN)制定的结直肠癌筛查测试策略指南,请单击这里here(免费注册,但需要登录)。理想状况下,需要逐步进行测试。
I. 肿瘤组织的检测 (Table 2)
MSI和IHC允许筛选肿瘤组织以帮助识别可能患有Lynch综合征的个体。这些方法可以通过各种策略实现:
方法1. 使用MSI和/或IHC测试对所有的结直肠癌进行筛查[EGAPP 2009]。
- 研究表明,这种策略越来越多地被学术医疗中心采用,社区医院应用的范围小一些 [Beamer et al 2012]。
- 由于Lynch综合征子宫内膜癌的比例和结直肠癌相似,因此Lynch综合征的普查已经扩展到所有新诊断的子宫内膜癌[Resnick et al 2009, Moline et al 2013]。
- 这些方法识别具有Lynch综合征的个体和家庭的成功率,在很大程度上取决于对异常结果进行跟踪,并确保遵循适当的遗传咨询和测试的系统 [Heald et al 2013]。
- Lynch综合征筛查网络Lynch Syndrome Screening Network的建立是为了帮助制定普遍筛查的最佳实践方法,并收集有关这些计划成果的长期数据。
注意:虽然IHC和MSI测试肿瘤组织的组合方法是理想的,但单独使用IHC测试对于大规模筛查程序可能更为可行,因为它比MSI测试更容易获得,并提供有关哪个MMR基因最有可能含有胚系 致病性变异。例如,对于表现出MLH1和PMS2表达缺失的肿瘤,MLH1启动子甲基化和BRAF V600E变体的肿瘤组织测试可以帮助区分散发性肿瘤和更可能具有胚系MLH1致病性变异的肿瘤。
方法2. 针对患有结直肠癌或其他Lynch综合征相关癌症个体的肿瘤组织IHC和/或MSI测试以及表明诊断的其他特征,例如肿瘤发病年龄早或强烈的家族史。
- 相关的研究还在继续,以便确定可能有助于区分结直肠癌和子宫内膜癌个体子集的因素,使得筛查Lynch综合征的特异性更高,从而达到筛查计划成本的最小化。
- 建立贝塞斯达指南是为了帮助识别可能患有Lynch综合征的人,然后进一步行肿瘤组织的MSI/IHC检测[Umar et al 2004]。虽然他们在鉴定患有Lynch综合征个体时的敏感性高达94%,但他们的特异性仅为25%[Syngal et al 2000]。
- 发病年龄和病理特征也被用于预测哪些个体最有可能具有MMR基因的致病性变异 [Rabban et al 2014]。
II. 分子遗传学检测 (Table 3)
方法 1. 根据肿瘤检测结果推荐 (Table 2)
方法 2. 根据以下风险评估模型之一,有助于预测Lynch综合征相关基因之一存在胚系 致病性变异的可能性:
- PREMM1,2, 6 Model 基于4539个人通过商业实验室进行MSH2、MLH1和MSH6基因检测的数据。该模型包含先证者、一级亲属和二级亲属的Lynch综合征相关癌症(结肠癌、子宫内膜癌、胃癌、卵巢癌、小肠癌、泌尿道/肾脏癌、胆管癌、胶质母细胞瘤、皮脂腺肿瘤和胰腺癌)的信息和发病年龄。来自MSI和IHC测试的数据不包括在内。基于基因型/表型数据,该模型提供了每个MMR基因中检测致病性变异的可能性。使用5%突变概率作为MMR测试的标准,该模型估计的敏感性 为90%,特异性为54%[Kastrinos et al 2011]。该模型还能够使用家族史来估计未患癌症的人拥有MMR 基因致病性变异的可能性。
- MMRpredict来自基于人群的队列研究的数据,该队列在55岁之前被诊断患有结直肠癌,他们都进行了MLH1、MSH2和MSH6致病性变体的测试。纳入了来自MSI和IHC测试的数据,以及一级亲属中结直肠癌和/或子宫内膜癌的数据。该模型只能用于评估受累的个体;尚不清楚该模型对老年人肿瘤患者的准确性 [Barnetson et al 2006]。
- MMRpro(参见CancerGene [需要注册])包含有关 先证者、一级亲属和二级亲属的结直肠癌和子宫内膜癌的信息、发病年龄,以及IHC和MSI检测的结果,以估计一个家庭中出现MLH1、MSH2或MSH6的 胚系 致病性变异的可能性;使用孟德尔模型来确定家庭成员可能遗传了种系致病变异;并计算个体患结肠癌和子宫内膜癌的风险。
- 致病性变异数据来自文献中报道的基于临床和人群的数据;癌症风险评估基于对五项大型Lynch综合征研究的荟萃分析的外显率 数据。由于该模型未包含大多数结肠癌以外的其它肿瘤的信息,因此尚不清楚它是否能够准确识别最可能发生突变的基因 [Chen et al 2006]。
方法 3. 在符合阿姆斯特丹标准的家庭中放弃MSI测试,直接进行胚系 分子遗传学检测。由于二代测序next-generation sequencing降低了测试所有MMR基因和EPCAM的成本,因此这种方法变得更具成本效益。然而,由于MMR表达缺失的结肠癌家族与MMR表达正常的癌症家族之间的癌症风险估计存在显著差异,肿瘤组织通常仍然是综合评估的一部分[Bapat et al 2009]。在以下情况下可以考虑直接行MMR基因的胚系基因测试。
当没有肿瘤组织可用于肿瘤检测时:
- 分子检测应该从被诊断患有Lynch综合征相关癌症的家庭成员开始。
- 对所有四种MMR基因和EPCAM进行分子检测。测试可以以逐步的方式进行,从最可能存在致病性变异的基因开始。
- 另一种策略是使用表型靶向检测,同时评估所有MMR基因。注意:结肠癌的基因套餐可能包括与Lynch综合征无关的其他基因。
- 表型靶向检测的使用可以在不能进行肿瘤测试时进行,以帮助缩小所涉及的基因,从而快速鉴定出致病性变异。
- 在没有关于肿瘤MSI状态的情况下,不可能区分Lynch综合征和家族性结直肠癌的其他原因。
当没有肿瘤组织或活着的受累的亲属可用于测试时:
- 成本效益分析表明,在这种情况下测试未受影响的个体是值得的,因为如果确定了引起Lynch综合征的胚系变异,家庭将获得显著的益处 [Dinh et al 2011]。
临床特征
临床描述
Lynch综合征是由于错配碱基修复 基因 的胚系 致病性变异 引起的,或者与肿瘤的MSI相关,他们患结肠癌和其他癌症的风险增加,包括子宫内膜癌、卵巢癌、胃癌、小肠癌、肝胆管癌、上尿路癌、脑和皮肤肿瘤(Table 4).。
表4. Lynch综合征患者和一般人群70岁以前的患癌风险
| 肿瘤类型 | 普通人群的风险 | 林奇综合征 (MLH1和MSH2 杂合性变异) | |
|---|---|---|---|
| 风险 | 平均发病年龄 | ||
| 结肠 | 4.8% | 52%-82% | 44-61岁 |
| 子宫内膜 | 2.7% | 25%-60% | 48-62岁 |
| 胃 | <1% | 6%-13% | 56岁 |
| 卵巢 | 1.4% | 4%-12% | 42.5岁 |
| 肝胆管 | <1% | 1.4%-4% | 未报道 |
| 泌尿管道 | <1% | 1%-4% | ~55岁 |
| 小肠 | <1% | 3%-6% | 49岁 |
| 大脑/中枢神经系统 | <1% | 1%-3% | ~50岁 |
| 皮脂腺肿瘤 | <1% | 1%-9% | 未报道 |
结肠癌。 基于符合阿姆斯特丹标准的高风险家庭的初步研究发现,结肠癌的终身风险高达82%,平均发病年龄为44岁。这些癌症的三分之二发生在近端结肠[Lynch et al 1993, Lynch & Smyrk 1996, Aarnio et al 1999]。
最近对MLH1和MSH2 致病性变异杂合子的研究报道的患癌风险较以前低一些,男性的风险为66%-69%,女性的风险为43%-52%,平均诊断年龄为61岁 [Hampel et al 2005, Stoffel et al 2009]。
对于MSH6和PMS2 致病性变异的杂合子,结肠癌风险的估计值也较低。已经发现,对于MSH6致病性变异体,男性和女性杂合子80岁以前患结肠癌的风险分别为44%和20%。虽然远低于MLH1和MHS2致病性变异体的风险,但这仍然代表结肠癌风险的八倍增加[Baglietto et al 2010]。对于PMS2致病性变异体杂合子,70岁之前患结肠癌的风险为15%-20%[Senter et al 2008]。
有关EPCAM致病性变异 患者的癌症风险数据仍然有限,但Kempers et al [2011] 报道了194名患有EPCAM 缺失的患者的研究结果。从这个队列中,他们估计到70岁时,结直肠癌的累积发病率为75%(95%CI 65-86%)。结肠癌的风险在仅具有EPCAM基因3'端缺失的病人,和同时包含EPCAM和MSH2缺失的病人之间没有差异。
在匹配肿瘤的分期之后,Lynch综合征相关的结肠癌患者的预后好于散发性结肠癌[Watson et al 1998]。这是一个意外的发现,因为Lynch综合征相关结肠癌的分化程度低,而分化程度低的患者通常预后不良。 Lynch综合征相关结肠癌的组织学特征包括:分化差、淋巴细胞浸润肿瘤上皮、粘液腺癌、印戒细胞癌或筛孔状组织学特征。
子宫内膜癌。患有Lynch综合征的女性患子宫内膜癌的终生风险为25%-60%,这使其成为Lynch综合征中第二常见的癌症 [Aarnio et al 1999, Vasen et al 2001, Hampel et al 2005, Stoffel et al 2008]。基于高危家庭的研究发现平均诊断年龄约为48岁;基于人群的研究显示,诊断年龄较晚(62岁)[Vasen et al 1994, Hampel et al 2005]。
虽然MSH6的突变主要是中度增加结肠癌的风险,但是MSH6杂合子还具有44%的子宫内膜癌的风险,类似于报道的MLH1和MSH2杂合子的风险水平 [Baglietto et al 2010]。在患有结肠癌和子宫内膜癌的Lynch综合征女性中,约50%首先患有子宫内膜癌 [Watson et al 1994, Lu et al 2005]。首次发生结肠癌的Lynch综合征女性之后再患子宫内膜癌的风险,在首次诊断结肠癌之后的10年内估计为26% [Obermair et al 2010]。
Kemper等人发现70岁时子宫内膜癌的累积风险为12%(95%CI :0-27%)。同时累及EPCAM和MSH2的缺失变异女性的患癌风险与MSH2致病性变异女性相似[Kempers et al 2011]。
总体而言,Lynch综合征占所有子宫内膜癌的约2%[Hampel et al 2006]。在Lynch综合征相关的子宫内膜癌中已经报道了类似于Lynch综合征相关结肠癌的生存优势[Maxwell et al 2001]。
胃癌。 对于MLH1或MSH2致病性变异的杂合子中胃癌的估计风险为6%-13%。具有MSH2致病性变异的男性患病风险最大 [Watson et al 2008, Capelle et al 2010]。在患有其他胃癌危险因素的国家,例如幽门螺杆菌感染的高发病率,已经报道了更高的风险 [Park et al 2000]。胃癌的平均诊断年龄为56岁。肠型腺癌是Lynch综合征相关胃癌最常见的病理学 [Aarnio et al 1997],组织学上与hereditary diffuse gastric cancer(弥漫性胃癌)不同,弥漫性胃癌最常见于由CDH1致病性变异引起的遗传性弥漫性胃癌 [Guilford et al 1999]。然而,Capelle等报道,高达20%的Lynch综合征相关胃癌可能是弥漫型 [Capelle et al 2010]。
卵巢癌。 卵巢癌的风险因发生胚系突变的MMR基因而异。已经发现MLH1致病性变异 杂合子的风险为4%-6%,而MSH2杂合子具有8%-11%的风险。 Lynch综合征相关卵巢癌的平均诊断年龄为42.5岁;然而,也有在很小的年龄就被诊断为卵巢癌的病例报道。大约30%的Lynch综合征相关卵巢癌在35岁之前被诊断出 [Watson et al 2008]。
病理类型的分布类似于散发性卵巢癌。交界性卵巢肿瘤似乎与Lynch综合征无关 [Watson et al 2001]。
其它癌症。 其他具有某些特征的Lynch综合征相关癌症也有报道:
- 泌尿系统癌症。最常见的与Lynch综合征相关的泌尿系统癌是输尿管和肾盂的移行细胞癌。
最近的一项研究表明,Lynch综合征患膀胱癌的风险可能也会增加。一项来自荷兰的关于Lynch综合征的队列研究表明,男性患膀胱癌的相对风险为4.4,女性为2.2。那些检测了MSI和/或IHC的研究结果显示,大多数肿瘤组织表现出MSI和/或具有胚系变异基因相应蛋白质的表达缺失 [van der Post et al 2010]。
对曾患结直肠癌的Lynch综合征患者的另一项研究也显示膀胱癌(7.22,95%CI = 4.08-10.99)和其他泌尿系统癌症(肾、肾盂和输尿管)的风险增加(12.54,95%CI 7.97-17.94)。这些额外的证据表明,膀胱癌应该包括在Lynch综合征癌症谱中 [Win et al 2013]。
尿路癌的风险因性别和突变基因的不同而显著不同,在男性杂合的MSH2致病性变异中存在较高的风险。具有MLH1致病性变异的女性具有约1%的风险,而具有MSH2致病性变异的男性估计风险高达27%[Watson et al 2008]。 - 小肠癌。 小肠癌的风险估计在3%至6%之间 [Watson et al 2008]。十二指肠和空肠是小肠癌最常见的部位,50%的可以通过上消化道内窥镜检查发现[Schulmann et al 2005]。大多数小肠癌是腺癌 [Rodriguez-Bigas et al 1998, Schulmann et al 2005]。
- 胰腺癌。 Kastrinos et al [2009] 根据报道的家族史进行的一项研究发现,70岁之前,胰腺癌患者的风险增加了8.6倍 [Kastrinos et al 2009]。一项纳入了446名患有MMR致病性变异的患者和1029名亲属的前瞻性研究发现,在中位随访时间为5年的随访期内,发现胰腺癌的风险增加(SIR,10.68; 95%CI 2.68-47.70),不具有致病性变异的个体的患癌风险并未增加[Win et al 2012]。然而,其他研究并未证明风险增加[Barrow et al 2009]。已发现Lynch综合征是家族性胰腺癌的罕见原因 [Gargiulo et al 2009]。
- 脑肿瘤。脑瘤的风险估计约为2%[Schulmann et al 2005]。然而,Barrow et al [2009] 估计患有双等位基因的MSH2致病性变异体的人患病风险增加了16倍。最常见的中枢神经系统肿瘤类型是胶质母细胞瘤 [Hamilton et al 1995, Wimmer & Etzler 2008]。
- 所描述的皮肤皮脂腺肿瘤包括:皮脂腺瘤、皮脂腺上皮瘤、皮脂腺癌和角化棘皮瘤[Schwartz & Torre 1995, Misago & Narisawa 2000]。与Lynch综合征相关的皮脂腺肿瘤表现出MSI [Entius et al 2000, Machin et al 2002]。 Lynch综合征患者皮脂腺肿瘤的发病率的数据有限。研究发现,在MMR基因中具有胚系 致病性变异的个体中有1%至9%具有皮脂腺肿瘤[Ponti et al 2006, South et al 2008]。
- 前列腺癌。 一些研究表明Lynch综合征与前列腺癌有关,风险增加2-5倍 [Raymond et al 2013b, Haraldsdottir et al 2014, Ryan et al 2014]。 Raymond et al [2013b]发现,在60岁之前拥有MMR致病性变异的男性患前列腺癌的风险增加[Raymond et al 2013b],而Haraldsdottir et al [2014] 的分析发现,具有MMR致病性变异体的个体中发生前列腺癌的年龄更早,并且更具侵袭性表型。
- 乳腺癌。乳腺癌和Lynch综合征之间的关系尚未解决。一项系统评价纳入了21项研究,13项研究未证实Lynch综合征患者患乳腺癌的风险增加,8项研究证实风险增加[Win et al 2013]。迄今为止,仅在一项前瞻性研究中评估了乳腺癌风险。发现具有致病性变异的个体的乳腺癌标准发病风险比为3.95(95%CI 1.59-8.13),乳腺癌诊断的中位年龄为56岁。 Walsh et al [2010]的一项研究表明,在MMR基因中具有致病性变异的个体中51%的乳腺癌组织表现出胚系突变基因对应蛋白质的免疫组织化学染色缺失。由于一般人群中乳腺癌的高频率,散发性乳腺癌的存在使得乳腺癌与Lynch综合征的关联分析复杂化。
已经报道了具有MMR致病性变体的个体有可能发生几种类型的肉瘤,包括纤维组织细胞瘤、横纹肌肉瘤、平滑肌肉瘤和脂肪肉瘤 [Sijmons et al 2000, den Bakker et al 2003, Nilbert et al 2009]。 Nilbert et al [2009] 确定Lynch综合征患者中8例肉瘤中有6例表现出MMR缺陷,这表明肉瘤也可能是Lynch综合征肿瘤谱的一部分。由于肉瘤的罕见性,可能难以确定其与Lynch综合征相关的风险程度。
在患有Lynch综合征的家庭中也报道了肾上腺皮质癌(ACC)。密歇根大学遗传性癌症诊所进行的这项最大的关联研究发现,114名(1.7%)ACC患者中有两名患有与Lynch综合征一致的家族史,并且确定了MMR致病性变异。通过135例具有致病性MMR变异的个体的病例回顾进一步评估该关联,其鉴定了两个人也有ACC(1.4%)[Raymond et al 2013a]。
Lynch综合征变种
- Muir-Torre综合征 的定义是皮肤的皮脂腺肿瘤和一种或多种内部脏器恶性肿瘤的组合,通常见于Lynch综合征。所描述的皮脂腺肿瘤的类型包括:皮脂腺瘤、皮脂腺上皮瘤、皮脂腺癌和角化棘皮瘤[Schwartz & Torre 1995, Misago & Narisawa 2000]。
- Turcot综合征被定义为结直肠癌或结直肠腺瘤,合并中枢神经系统的肿瘤。临床表现从大量结肠息肉到单一息肉或结直肠癌不等。
Turcot综合征通常由APC的致病性变异 (参见APC-Associated Polyposis Conditions)或由与Lynch综合征相关的错配碱基修复基因之一的致病性变异引起 [Hamilton et al 1995]。患有APC致病性变异的个体通常具有更多的息肉;然而,息肉数量的显著重叠存在于由APC致病性变异体引起的Turcot综合征和由错配修复基因中的致病性变异引起的Turcot综合征的个体之间[Hamilton et al 1995]。 中枢神经系统肿瘤的病理学可以帮助区分潜在的遗传原因:APC致病性变异更常与髓母细胞瘤相关;错配修复基因中的致病性变异更常与胶质母细胞瘤相关。
与错配修复基因突变相关的脑肿瘤表现出MSI [Hamilton et al 1995, Suzui et al 1998]。 - 纯合性错配碱基修复变异体 目前已有关于MLH1、MSH2、MSH6和PMS2中的纯合性致病性变异的罕见病例的报道。受影响的个体通常在20岁之前发生结肠癌或小肠癌。据报道,三分之一的拥有MMR基因的双等位基因的致病性变异的儿童有超过十个息肉。还有血液学肿瘤、脑肿瘤和咖啡-斑点斑的报道[Wimmer & Etzler 2008, Durno et al 2010, Bakry et al 2014]。受累个体的皮肤表型可能与neurofibromatosis type I(I型神经纤维瘤病)中的皮肤表型非常相似 [Wimmer 2012]。然而,更有可能是,导致Lynch综合征的一个基因中的纯合致病性变异或复合杂合的个体,具有以下一种或多种表现:
- 综合征的家族史;
- 父母有血缘关系;
- 至少有一位家长有Lynch综合征的临床表现。
基因型 - 表型相关性
四种MMR基因的癌症风险各不相同。
MSH2基因的杂合性致病性变异的患者发生结肠外癌症的风险最大;对于具有MSH6、PMS2或EPCAM基因的杂合性致病性变异的患者,结肠外癌症(除子宫内膜癌)的风险较低。
在具有Lynch综合征的Muir-Torre变体的个体中,已报道MSH2致病性变异比其他三个MMR基因中的致病性变异更常见[South et al 2008]。
MSH6的杂合性致病性变异与低度微卫星不稳定(MSI-L)的肿瘤相关。与其他MMR基因引起的Lynch综合征相比,具有MSH6致病性变异的家族中的癌症可能发病更晚,而且结肠癌通常位于结肠的更远端。子宫内膜癌通常在患有MSH6致病性变异的女性中出现[Wu et al 1999, Berends et al 2002]。具有MSH6致病性变异的家族,与具有MLH1或MSH2致病性变异的家族相比,结肠癌的风险略低,而子宫内膜癌的风险更高[Berends et al 2002, Baglietto et al 2010]。
PMS2的杂合性致病性变异的患者发生任何Lynch综合征相关癌症的风险都是最低的(25%-32%的风险) [Senter et al 2008]。
导致MSH2表观遗传沉默的EPCAM缺失与结肠癌风险显着增加有关。研究表明,结肠干细胞中EPCAM的表达水平很高;然而,由于EPCAM在所有组织中均未高水平表达,因此EPCAM对结肠外癌症风险的影响尚不清楚。Kempers et al [2011]报道,与MMR基因突变相比,EPCAM缺失患者的子宫内膜癌风险较低。结肠外癌症的风险也取决于缺失的程度。 EPCAM的3'端缺失已被证明可以降低结肠外癌症的风险,而累及MSH2的缺失会产生类似于MSH2基因内致病性变异的结肠外癌症的风险[Tutlewska et al 2013]。
预期发病年龄
一项研究报告了Lynch综合征中的遗传早现(即,后代的发病年龄比父母年轻) [Westphalen et al 2005],但是尚未被证实。另一项研究中,后代的发病年龄比父母年龄小,这一研究结果是由于出生队列偏倚引起的 [Tsai et al 1997]。
命名
Lynch综合征也被称为遗传性非息肉病性结直肠癌(HNPCC)。在遗传性结肠癌领域工作的临床医生和研究人员建议恢复使用原名--Lynch综合征,来指定具有MMR缺陷的个体和家庭,并将其与其他形式的家族性结肠癌区别开来 [Bellizzi & Frankel 2009, Weissman et al 2011]。
发病率
Lynch综合征约占结肠癌的1%-3%,占子宫内膜癌的0.8%-1.4% [Kowalski et al 1997, Chadwick et al 2001, Cunningham et al 2001]。
Lynch综合征的人群患病率估计为1:440 [Chen et al 2006]。
遗传相关(等位)疾病
除GeneReview中讨论的那些表型之外,没有发现与MLH1、MSH2、MSH6、PMS2和EPCAM的致病性变异有关的其他表型。
鉴别诊断
家族性结直肠癌。 评估肿瘤组织的MSI状态对于区分Lynch综合征与家族性结直肠癌是重要的。在对肿瘤MSI稳定的、有强烈的结直肠癌家族史的家族进行的研究中,没有发现错配碱基修复 基因的胚系 致病性变异所伴随的结肠外癌症风险的增加。在具有MSI稳定肿瘤的家族中,结直肠癌的风险似乎也较低 [Abdel-Rahman et al 2005, Lindor et al 2005, Mueller-Koch et al 2005]。许多候选基因、低外显率等位基因和环境风险因子也与家族性结直肠癌有关。
轻表型familial adenomatous polyposis(轻表型家族性腺瘤性息肉病,AFAP)。 FAP的这种较温和的表现,也是由APC中的致病性变异引起的,其特征在于比传统FAP息肉更少和发病年龄更晚。在AFAP中,通常观察到少于100枚息肉。胃底和十二指肠也有息肉;然而,在AFAP中可能不存在FAP中常见的许多结肠外表现(例如:表皮囊肿、牙齿异常、先天的视网膜色素上皮肥大、硬纤维瘤)。与AFAP相关的息肉和结肠癌通常不表现出MSI。AFAP以常染色体显性遗传的方式遗传。
APCp.Ile1307Lys 的致病性变异。这种致病性错义变异与经典的FAP表型无关;然而,在APC中具有p.Ile1307Lys致病性变异的个体患结肠癌的风险增加约两倍。该变异在具有Ashkenazi Jewish(德系犹太人)血统的人群中发生率约有6%Laken et al 1997]。有关此变异体的信息,请参阅APC-Associated Polyposis Conditions (APC相关息肉病)。
MUTYH-associated polyposis(MUTYH相关的息肉病)。已经在具有多个腺瘤性息肉的个体中描述了MUTYH(MYH)中的致病性变异。已经在以下个体中检测到了MUTYH的致病性变异:(1)约30%的具有15-100枚息肉的个体; (2)一小部分具有经典FAP表型且没有可识别的APC致病性变异的个体; (3)在没有多发性息肉的情况下有结肠癌家族史的个体 [Jo et al 2005]。以常染色体隐性遗传的方式遗传 [Sieber et al 2003]。
错构瘤性息肉病综合症。 与错构瘤性息肉和结肠癌风险增加相关的几种情况,通常可以通过其结肠外表现以及息肉的病理性质(错构瘤性息肉vs.腺瘤性息肉)来区分:
- Juvenile polyposis syndrome (幼年性息肉病综合征,JPS), 由SMADA4和BMPR1A中的致病性变异引起。
- Peutz-Jeghers syndrome (黑斑息肉病综合征,PJS), 由STK11的致病性变异引起。
- PTEN hamartomatous syndromes (PTEN错构瘤综合征,包括Cowden综合征,Bannayan-Riley-Ruvalcaba [BRR]综合征), 由PTEN基因的致病性变异引起。
Hereditary diffuse gastric cancer(遗传性弥漫性胃癌,HDGC). 由CDH1的致病性变异引起的胃癌,通常是腺癌。
BRCA1/BRCA2 hereditary breast/ovarian syndrome(BRCA1 / BRCA2遗传性乳腺癌/卵巢癌综合征) :在评估具有包括卵巢癌的癌症家族史的个体时,应考虑BRCA1 / BRCA2遗传性乳腺癌/卵巢癌综合征。
管理
初步诊断后的评估
为了确定被诊断患有Lynch综合征个体的疾病程度和需求,建议要进行全面的评估(在随访监测 Surveillance中做了总结)。
各种临床表现的治疗
Lynch综合征相关结肠癌的治疗。如果检测到结肠癌,建议采用全结肠切除术+回肠直肠吻合术,而不是节段性/部分结肠切除术,因为异时性癌症的风险很高[Lynch et al 1988, Aarnio et al 1995, Church & Simmang 2003]。对符合阿姆斯特丹标准的家庭的296名个体(253名行部分结肠切除术,43名行全结肠切除术)的研究发现,中位随访104个月,22%的部分结肠切除患者发生了高风险的腺瘤和25%的患者发生了第二次原发性结肠癌,而对照组只有11%和8%的患者发生了腺瘤和结肠癌[Kalady et al 2010]。注意:由于Lynch综合征的诊断通常在治疗初始癌症之后才被考虑,因此许多被诊断患有Lynch综合征的个体之前已经通过节段性的结肠切除术治疗了癌症。
虽然时间上可能很困难,但通过MSI和IHC评估肿瘤活检标本(以及,如果有需要,MHL1启动子甲基化状态)可能有助于确定最佳手术方法。一项研究表明,结肠癌患者在诊断时更容易接受基因检测[Porteous et al 2003]。
在Lynch综合征中出现的其他肿瘤与一般人群一样进行管理。
主要表现的预防
在生育结束后,可以考虑预防性切除子宫和卵巢(在癌症发生之前)。
由于常规结肠镜检查是结肠癌的有效预防措施,因此一般不建议对Lynch综合征患者进行预防性结肠切除术(癌症发生前切除结肠)。
监测
结肠癌。定期行结肠镜检查并切除尚未癌变的息肉可降低Lynch综合征患者结肠癌的发病率。 2009年一项芬兰的队列研究(该研究对筛查的依从性很高),发现Lynch综合征患者的死亡率与其突变阴性亲属相比并未增加,这表明每年行结肠镜检查有助于预防和检测结肠癌[Järvinen et al 2009]。在这项研究中,四个人被诊断患有结肠癌和淋巴结转移:一个在基线结肠镜检查中被诊断出来;其他三人在最后一次结肠镜检查后两年多被诊断出来。因此,目前的建议是每隔1-2年进行一次结肠镜检查,从20至25岁开始,或者在家庭中最早诊断之前的2-5年,以较早者为准 [Järvinen et al 2009, Engel et al 2010, NCCN 2013]。
在MSH6和PMS2杂合子中,结肠癌的风险较低;因此,开始行结肠镜检查的时间可延迟到30岁 [Senter et al 2008, Baglietto et al 2010]。
注意:建议进行全结肠镜检查而不是软式乙状结肠镜检查,因为Lynch综合征中近端结肠癌占多数 [Lynch & Smyrk 1996]。
子宫内膜癌。子宫内膜癌监测的结果不如结肠癌。
因为许多子宫内膜癌可以根据症状在早期诊断,所以应该对女性患者进行教育,告知她们子宫内膜癌有哪些症状。
目前,国家综合癌症网络(NCCN)不建议对子宫内膜癌或卵巢癌进行任何特异性筛查[NCCN 2013]。
关于经阴道超声检查和子宫内膜活检的有效性的研究已经产生了相互矛盾的结果。需要进一步研究以确定经阴道超声检查和子宫内膜活检的组合能否在早期检测出子宫内膜癌。
- 在使用经阴道超声检查筛查子宫内膜癌的研究中,没有检测到癌症;然而,在研究过程中根据症状表现检测到2例癌症[Dove-Edwin et al 2002]。
- 芬兰的队列研究发现,每2-3年进行一次子宫内膜活检和经阴道超声检查,可以诊断早期癌症。然而,由于子宫内膜癌在早期阶段经常出现症状,因此,尚不清楚筛查能否改善检出率[Järvinen et al 2009]。
卵巢癌。没有针对Lynch综合征女性的特定的卵巢癌筛查试验。值得注意的是,使用CA-125血液检查和经阴道超声检查筛查卵巢癌在其他高风险人群中无效,如BRCA1或BRCA2致病性变异 [Evans et al 2009]。
胃癌和十二指肠癌。 上消化道内窥镜检查可用于筛查胃和十二指肠的癌症。目前,NCCN建议从30至35岁开始行上消化道内窥镜检查(带侧视功能+十二指肠镜检查),并根据检查结果每3-5年重复一次。那些有慢性炎症、萎缩性胃病和/或肠上皮化生证据的人需要更频繁的检查。注意:应对活检组织进行幽门螺杆菌感染的评估,以便进行适当的治疗[NCCN 2013]。
关于在Lynch综合征中早期检测胃癌的上消化道内窥镜检查有效性的数据是有限的。
- 一项研究表明,由于缺乏可识别的前驱病变,因此胃癌筛查无益 [Renkonen-Sinisalo et al 2002]。
- 一项来自荷兰的关于胃癌风险的研究表明,风险水平足以进行筛查;然而,由于87%的癌症发生在45岁以后,因此从45岁开始筛查可能是最具成本效益的[Capelle et al 2010]。
- Schulmann et al [2005] 发现,Lynch综合征患者中约有50%的小肠癌位于十二指肠,这表明上消化道内镜可能对筛查有用。然而,目前还没有研究证实了上消化道内镜检查筛查十二指肠癌的功效。
远端小肠。此时,关于筛查远端小肠中癌症的数据是有限的。胶囊内窥镜检查和小肠造影可用于评估小肠,但目前不建议常规使用这些方法进行小肠筛查,尽管它们可能有助于评估有症状的个体。
泌尿系统。NCCN建议考虑从25岁-30岁开始每年行尿常规检查[NCCN 2013]。
其他癌症。 目前,尚无针对其他Lynch综合征相关癌症的特异性筛查建议。应鼓励受影响的个体遵循其他一般人群筛查指南,并就健康状况的变化或持续性症状及时寻求医疗帮助。
要避免的药物/环境
吸烟会增加Lynch综合征患结直肠癌的风险[Watson et al 2004, Pande et al 2010]。
亲属的风险评估
早期识别与Lynch综合征相关的癌症可能允许及时干预并改善最终结果;因此,对无症状的高危亲属进行监测,并及时发现他们的早期表现是适当的。
当在Lynch综合征家族中发现MMR基因的致病性变异时,应向所有一级亲属(父母、兄弟姐妹和子女)提供致病性变异的分子遗传学检测 。
一般而言,对于年龄小于18岁的高危人群,不推荐行Lynch综合征的分子遗传学检测。然而,由于有部分Lynch综合征患者,在很小的年龄就被诊断出癌症 [Huang et al 2001],因此建议在家族最早发病年龄之前十年开始筛查。在这种情况下,在开始癌症筛查之前,为年龄小于18岁的儿童提供分子遗传学检测是合适的。
有关高危亲属进行检测的遗传咨询目的的相关问题,请参阅Genetic Counseling。
怀孕管理
理想情况下,合并Lynch综合征的孕妇应当在怀孕期间进行癌症筛查。在尝试怀孕之前,鼓励受累的女性在将她的癌症筛查全部更新。如果Lynch综合征妇女在怀孕期间被诊断患有癌症,应向她提供有关癌症治疗方案及其对胎儿潜在影响的咨询。
正处于研究当中的检查和治疗方法
染色内镜和强化结肠镜检查。 2008年的一项研究比较了染色内镜检查和强化结肠镜检查在早期发现Lynch综合征患者息肉方面的有效性。在标准结肠镜检查后,研究参与者随机接受第二次结肠镜检查,其中使用染色内镜检查或第二次结肠镜检查并仔细检查。这项研究发现,尽管在标准结肠镜检查中经常会遗漏息肉,但是通过染色内镜检查和仔细观察第二次结肠镜检查发现的额外息肉数量之间没有差异 [Stoffel et al 2008]。需要进一步的研究来确定新的结肠镜检查技术是否为Lynch综合征患者的筛查提供了额外的益处。
化学预防研究
- 在1071名患有Lynch综合征的个体中进行了一项比较安慰剂与阿司匹林和安慰剂与抗性淀粉的四臂试验。在阿司匹林或抗性淀粉干预组中没有观察到对结直肠肿瘤风险的影响[Burn et al 2008]。然而,当这个队列被跟踪十年后,接受阿司匹林的组确实显示结肠癌发病率降低了63%[Burt 2012]。未发现抗性淀粉对结肠癌风险有影响[Mathers et al 2012]。
- 虽然口服避孕药可降低一般风险人群的女性患子宫内膜癌和卵巢癌的风险,但尚不清楚它们是否能为Lynch综合征女性患者带来同样的益处。
可在ClinicalTrials.gov 中搜索有关各种疾病和病症的临床研究信息。
遗传咨询
遗传咨询是向个人和家庭提供有关遗传疾病的性质、遗传和影响的信息的过程,以帮助他们做出明智的医疗和个人决定。以下部分主要涉及的是,遗传风险评估,以及使用家族史和基因检测来阐明家庭成员的遗传状况。本节不是为了解决个人可能面临的所有个人、文化和伦理问题,也不能代替专业遗传人员的咨询。 -ED。
遗传的方式
Lynch综合征以常染色体显性遗传的方式遗传的。
家庭成员的风险
先证者的父母
- Lynch综合征的确切新突变率尚不清楚,但估计极低[Bisgaard & Bernstein 2003]。
先证者的同胞
- 应该向所有同胞提供家族中发现的致病性变异的分子遗传学检测。
先证者的后代。Lynch综合征患者的每个孩子有50%的机会继承这个致病性变异。
先证者的其他家庭成员。其他家庭成员的风险取决于他们与先证者的关系。家族史或分子遗传学检测可以帮助确定母亲或父亲的亲属是否处于危险之中。发现患有致病性变异或被诊断患有Lynch综合征相关癌症的家庭成员的后代可被认为具有50%的风险。对于有危险的人在没有患癌症的情况下死亡的家庭的后代,可以使用贝叶斯分析来帮助计算后代的风险。
相关的遗传咨询问题
有关高危亲属的评估,以及早期诊断和早期治疗的相关信息,请参阅Evaluation of Relatives at Risk(高危亲属的评估)和管理。
具体的风险问题。有几个因素可能会阻碍基于家族史的Lynch综合征的诊断。筛查和切除癌前息肉和预防性手术可预防一些高危亲属的结肠癌或子宫内膜癌;一些因其他原因死亡的年轻人可能从未患过癌症。
对具有明显de novo(新出现的)致病性变异的家庭的考虑。当Lynch综合征先证者的父母都没有Lynch综合征的致病性变异或临床证据时,先证者可能具有新出现的致病性变异。然而,MMR基因中的新出现的突变被认为是罕见的,并且还可以探索其他可能的非医学解释,包括非生物学父亲或母亲(例如,辅助生殖)或未公开的收养[Bisgaard & Bernstein 2003]。
家庭计划
- 确定遗传风险和产前检测可用性的讨论的最佳时间是在怀孕前。
遗传性癌症风险评估和咨询。 有关通过癌症风险评估识别有风险个体的医学、心理社会和伦理道德的全面描述,有或没有分子遗传学检测,请参阅癌症遗传风险评估和咨询 - 适用于卫生专业人员(Cancer Genetics Risk Assessment and Counseling – for health professionals)(PDQ的一部分®,国家癌症研究所)。
高危无症状成人的检测:使用分子遗传测试(Molecular Genetic Testing)中描述的技术可以测试Lynch综合征的高危无症状成人。此类检测无法预测症状是否会发生,或者如果发生,发病年龄、症状的严重程度和类型或疾病进展速度。当测试患有Lynch综合征的高风险个体时,应首先测试受累的家庭成员以确认该家族的分子诊断。
在做出基因检测决定之前,建议进行遗传咨询。Brain et al [2005]认为,对于有可治疗疾病风险的无症状成人,单一教育会议可能足以促进遗传咨询 。但是,预备信息可能有助于鼓励个人思考以前未考虑过的问题。遗传咨询包括讨论基因检测对个人和家庭成员的临床和心理社会影响。评估基因检测后的心理调整的研究尚未发现,获悉一个人患有Lynch综合征的致病性变异会导致不良的心理结果,或者临床痛苦的显著增加。然而,Lynch综合征患者的亚组,例如那些高级别的测试前痛苦、生活质量差或社会支持水平低的人,更容易发生心理疾病 [Vernon et al 1997, Dudok deWit et al 1998, Gritz et al 1999]。
年龄小于18岁的无症状个体的分子遗传学检测。一般而言,不建议对年龄小于18岁的高危人群进行Lynch综合征的基因检测。由美国医学遗传协会和美国人类遗传学会联合制定的指南规定,只有在影响其医疗管理的情况下,才应对18岁以下的个体进行预测性基因检测。建议将测试决定推迟到个人到达成年并且可以做出独立决定是,因为不推荐从20岁开始进行Lynch综合征相关癌症风险的管理。由于罕见报道的Lynch综合征患者在很小的年龄就被诊断出患有癌症 [Huang et al 2001],因此建议在家族最早发病年龄之前十年开始筛查。在一些家庭中,个人可能需要在18岁之前开始筛查。另见国家遗传咨询协会关于成人发病条件下未成年人基因检测的立场声明(position statement)和美国儿科学会和美国医学遗传学和基因组学政策声明(policy statement):基因检测和儿童筛查中的伦理和政策问题。
DNA储存 是指将DNA(通常从白细胞中提取)储存好,以备将来使用。DNA的存储因为测试方法和我们对基因、等位基因变异和疾病理解的不同,将来可能会有所改善,所以应该考虑为受累的个体储存DNA。
产前检查
如果确定了家族中的致病性变异,就可以通过实验室提供的感兴趣基因/疾病的测试,或定制化检测项目,以便对胎儿的患病风险进行产前诊断。
通常,对于那些成人发病而且有治疗方法的疾病(如Lynch综合征),产前检查的要求并不常见。医疗专业人员和家庭内部关于使用产前检查的观点可能存在差异,特别是如果考虑将检测用于终止妊娠而不是早期诊断。虽然大多数中心会考虑将产前检查的决定交给父母来选择,但对这些问题的讨论是必要的。
胚胎植入前遗传学诊断(PGD)可能是一些已确定致病性变异的家庭的一个选择。
资源
GeneReviews的工作人员选择了以下疾病特异性和/或orumbrella支持组织和/或登记处,以使这种疾病患者及其家人受益。 GeneReviews不对其他组织提供的信息负责。有关选择标准的信息,请单击此处 。
- Collaborative Group of the Americas on Inherited Colorectal Cancer (CGA)
- Hereditary Colon Cancer Takes Guts
- Lynch Syndrome InternationalP.O. Box 5456Vacaville CA 95688Phone: 707-689-5089Email: info@lynchcancers.com
- My46 Trait Profile
- National Cancer Institute (NCI)6116 Executive BoulevardSuite 300Bethesda MD 20892-8322Phone: 800-422-6237 (toll-free)Email: cancergovstaff@mail.nih.gov
- American Cancer Society (ACS)1599 Clifton Road NortheastAtlanta GA 30329-4251Phone: 800-227-2345 (toll-free 24/7); 866-228-4327 (toll-free 24/7 TTY)
- C3: Colorectal Cancer Coalition1414 Prince StreetSuite 204Alexandria VA 22314Phone: 877-427-2111 (toll-free); 703-548-1225Fax: 202-315-3871Email: info@fightcolorectalcancer.org
- Colon Cancer Alliance (CCA)1200 G Street NorthwestSuite 800Washington DC 20005Phone: 877-422-2030 (Toll-free Helpline); 202-434-8980Fax: 866-304-9075 (toll-free)
- Prospective Registry of MultiPlex Testing (PROMPT)PROMPT是一个针对经过多重基因检测、且发现可能与患癌风险增加有关的遗传变异的患者和家庭的在线研究登记网站。
分子遗传学
Molecular Genetics和OMIM表中的信息可能与GeneReview中的其他信息不同:表格可能包含更多最新信息。 - ED
表A. Lynch综合征: 基因和数据库
表B. Lynch综合征的OMIM入口 (View All in OMIM)
| 114500 | COLORECTAL CANCER; CRC |
| 120435 | LYNCH SYNDROME I |
| 120436 | MutL, E. COLI, HOMOLOG OF, 1; MLH1 |
| 158320 | MUIR-TORRE SYNDROME; MRTES |
| 185535 | EPITHELIAL CELLULAR ADHESION MOLECULE; EPCAM |
| 276300 | MISMATCH REPAIR CANCER SYNDROME; MMRCS |
| 600259 | POSTMEIOTIC SEGREGATION INCREASED, S. CEREVISIAE, 2; PMS2 |
| 600678 | MutS, E. COLI, HOMOLOG OF, 6; MSH6 |
| 609309 | MutS, E. COLI, HOMOLOG OF, 2; MSH2 |
| 609310 | COLORECTAL CANCER, HEREDITARY NONPOLYPOSIS, TYPE 2; HNPCC2 |
| 613244 | COLORECTAL CANCER, HEREDITARY NONPOLYPOSIS, TYPE 8; HNPCC8 |
分子遗传发病机制
Lynch综合征是由涉及错配碱基修复(MMR)途径基因的致病性变异引起的。该途径用于识别和去除单核苷酸错配或插入和缺失环。四种MMR基因的致病性变异可引起Lynch综合征[Peltomäki 2003]。错配修复基因的功能可以通过错义变异、截短变异、剪接位点变异、大片段缺失或基因组的重排来破坏。此外,EPCAM内的胚系缺失(不是MMR基因)可以通过使相邻MMR基因MSH2的失活来破坏MMR途径,即使MSH2本身未发生突变。
已报道Lynch综合征中癌症风险的遗传修饰因子:
- Zecevic et al [2006]发现在MMR基因中有一个致病性变异的患者中,较短的IGF1-CA重复与结肠癌风险增加(HR = 2.36; 95%CI = 1.28-4.36)和发病年龄早(44岁对56.5岁)有关。
- 据报道,RNASEL的变体与较早的发病年龄相关,Arg462纯合性变体的平均发病年龄为40岁,Gly462纯合性变体的平均年龄为34岁[Krüger et al 2005]。
- Pande et al [2008] 评估了许多基因,包括CYP1A1、EPHX1、GSTT1、GSTM1和GSTP1,发现CYP1A1中的两个序列变异可能与Lynch综合征患者结肠癌发病年龄早有关。
- 据报道,几种单核苷酸多态性与结直肠癌风险相关,但是这并未改变患有MMR致病性变异个体的癌症风险[Win et al 2013]。
MLH1的基因结构。MLH1的长度为57,357kb,其中19个编码外显子编码756个氨基酸的蛋白质。有关基因和蛋白质信息的详细摘要,请参见Table A,基因。
等位基因的致病性变异。据报道,MLH1有200多种不同的致病性变异 [Peltomäki 2003, Peltomäki & Vasen 2004];参见Table A。大片段缺失占胚系MLH1致病性变异的5%-10%。
MLH1的组成性甲基化失活,以及功能性等位基因的体细胞杂合性丢失,已被报道为Lynch综合征的罕见病因(~0.6%) [Niessen et al 2009]。由于启动子的甲基化和Lynch综合征的表型,这些个体在其整个组织中沉默了一个MLH1等位基因。大多数此类病例是单发的(即,一个家族中只出现一次),但已报道了一些遗传性高甲基化病例[Goel et al 2011]。这些病例无法通过MLH1的序列分析或大片段的重复/缺失分析检测到。
正常的基因产物。DNA错配碱基修复蛋白Mlh1与PMS2基因(PMS1蛋白同源物2)的产物二聚化,以协调与错配修复相关的其他蛋白质的结合,包括解螺旋酶、EXO1编码的蛋白质、增殖细胞核抗原(PCNA)、单链DNA结合蛋白(RPA)和DNA聚合酶 [Peltomäki 2003]。
异常的基因产物。MLH1在细胞水平以隐性方式起作用,其中肿瘤细胞中不存在功能性Mlh1蛋白。这是由于肿瘤中MLH1等位基因的失活导致的,这通常是由于失活突变或通过高甲基化使MLH1启动子沉默而发生的。
MSH2的基因结构。MSH2包含16个外显子,编码934个氨基酸的蛋白质。有关基因和蛋白质信息的详细摘要,请参见Table A,基因。
等位基因的致病性变异。在MSH2中已经鉴定出超过170种致病性变异[Peltomäki 2003, Peltomäki & Vasen 2004]。较高比例的Alu重复可能导致MSH2中基因组重排率高于MLH1[van der Klift et al 2005]。至少20%的MSH2胚系致病性变异是外显子或多克隆缺失。
正常的基因产物。DNA错配碱基修复蛋白MSH2(由MSH2编码的蛋白质)与DNA错配修复蛋白MSH6或MSH3形成异二聚体,并且用于识别配。已经提出滑动钳模型来描述异二聚体的结构。当夹钳沿DNA滑动时,认为DNA中的错配将被检测到[Fishel et al 1993, Gruber & Kohlmann 2003]。
异常的基因产物。MSH2在细胞水平以隐性方式起作用,其中肿瘤细胞中不存在功能性Msh2蛋白。这是由于肿瘤中MSH2两个等位基因的失活导致的,这通常通过杂合性丢失(LOH)的机制发生。已显示MSH2启动子甲基化,是在具有MSH2失活引起致病性变异的个体中沉默正常等位基因的失活事件。值得注意的是,这不是散发性结肠癌的常见原因。
MSH6的基因结构。MSH6包含10个外显子,编码1360个氨基酸的蛋白质。有关基因和蛋白质信息的详细摘要,请参见Table A,基因。
等位基因的致病性变异。MSH6中已发现30多种致病变异[Peltomäki & Vasen 2004]。外显子或多外显子缺失是胚系MSH6致病性变异的罕见原因。
正常的基因产物。由DNA错配碱基修复基因MSH6编码的蛋白质与DNA错配修复蛋白MSH2形成异二聚体,并通过滑动钳模型识别错配 [Fishel et al 1993, Gruber & Kohlmann 2003]。
异常的基因产物。MSH6在细胞水平以隐性方式起作用,其中肿瘤细胞中不存在功能性MSH6蛋白。这是由于肿瘤中MSH6双等位基因的失活导致的,这通常通过杂合性丢失(LOH)的机制发生。
PMS2的基因结构。PMS2包含15个外显子,编码862个氨基酸的蛋白质。已在7p22、7p12-13、7q11和7q22鉴定出多个假基因[Nicolaides et al 1995]。有关基因和蛋白质信息的详细摘要,请参见Table A,基因。
等位基因的致病性变异。PMS2中的胚系致病性变异是罕见的 [Hendriks et al 2006]。已经报道了单核苷酸变异和大片段基因重排。包括大片段缺失检测在内的研究发现,高达20%的致病性变异可能是大片段缺失 (Table 3)。由于许多假基因,PMS2的大片段缺失的检测在技术上是困难的,并且它对试图为整个基因提供全面的大片段缺失检测的实验室提出了重大挑战。目前可用的MLPA(多重连接依赖性探针扩增)试剂盒可以检测大片段缺失,但不能说明缺失是否存在于一种假基因中。与一组参考样本协同检测可以帮助确定缺失是否具有临床意义 [Vaughn et al 2011]。
异常的基因产物。PMS2在细胞水平以隐性方式起作用,其中肿瘤细胞中不存在功能性PMS2蛋白。这是由于肿瘤中PMS2双等位基因的失活导致的,这通常通过杂合性丢失(LOH)的机制发生。
EPCAM的基因结构。EPCAM包含9个外显子,编码314个氨基酸的蛋白质。有关基因和蛋白质信息的详细摘要,请参见Table A,基因。
等位基因的致病性变异。EPCAM的大片段缺失导致转录终止,是Lynch综合征家族的1%-2.8%的原因。其他不影响转录终止信号的EPCAM致病性变异引起常染色体隐性遗传的先天的 簇状肠病[Sivagnanam et al 2010]。
正常的基因产物。EPCAM在不同组织中表达水平不同。在结直肠干细胞中表达水平高,而在白细胞中表达水平低[Ligtenberg et al 2009]。关于EPCAM在大多数易患Lynch综合征相关癌症的组织中的表达情况知之甚少。
异常的基因产物。EPCAM缺失被认为是由Alu介导的重组事件引起的[Kuiper et al 2011]。消除EPCAM转录终止信号,将导致转录继续进入MSH2,并通过甲基化使MSH2启动子沉默。通过该机制,具有EPCAM缺失的顺式构型的MSH2等位基因在表达EPCAM的组织中失活,而其他MSH2等位基因不受影响。这些致病性变异以常染色体显性遗传的方式遗传,与MMR基因的胚系变异一样[Ligtenberg et al 2009]。
参考文献
发表的指南/共识声明
- American College of Medical Genetics technical standards and guidelines for genetic testing for inherited colorectal cancer (Lynch syndrome, familial adenomatous polyposis, and MYH-associated polyposis). Available online. 2014. Accessed 10-5-15. [PubMed: 24310308]
- American College of Medical Genetics/American Society of Human Genetics. Joint statement on genetic testing for colon cancer (pdf). Available online. 2000. Accessed 10-4-15.
- American Gastroenterological Association. Medical position statement: hereditary colorectal cancer and genetic testing (pdf). Available online. 2001. Accessed 10-5-15.
- American Society of Clinical Oncology. Expert statement: collection and use of a cancer family history for oncology providers. Available online. 2014. Accessed 10-5-15. [PMC free article: PMC3940540] [PubMed: 24493721]
- American Society of Clinical Oncology. Policy statement update: genetic testing for cancer susceptibility. Available online. 2010. Accessed 10-5-15.
- American Society of Colon and Rectal Surgeons. Practice parameters for the treatment of patients with dominantly inherited colorectal cancer (FAP and HNPCC). Available online. 2003. Accessed 10-5-15.
- Committee on Bioethics, Committee on Genetics, and American College of Medical Genetics and Genomics Social, Ethical, Legal Issues Committee. Ethical and policy issues in genetic testing and screening of children. Available online. 2013. Accessed 10-5-15. [PubMed: 23428972]
- Giardiello FM, Brensinger JD, Petersen GM. American Gastroenterological Association technical review on hereditary colorectal cancer and genetic testing (pdf). Available online. 2001. Accessed 10-5-15.
- National Comprehensive Cancer Network. Guidelines for colorectal cancer screening. Available online. Login required. 2013.
- National Society of Genetic Counselors. Position statement on genetic testing of minors for adult-onset disorders. Available online. 2012. Accessed 10-5-15.
- National Society of Genetic Counselors and the Collaborative Group of the Americas on Inherited Colorectal Cancer joint practice guideline. Identification of individuals at risk for Lynch syndrome using targeted evaluations and genetic testing. Available online. 2011. Accessed 10-5-15. [PubMed: 22167527]
引用文献
- Aarnio M, Mecklin JP, Aaltonen LA, Nystrom-Lahti M, Jarvinen HJ. Life-time risk of different cancers in hereditary non-polyposis colorectal cancer (HNPCC) syndrome. Int J Cancer. 1995;64:430 - 3. [PubMed: 8550246]
- Aarnio M, Salovaara R, Aaltonen LA, Mecklin JP, Jarvinen HJ. Features of gastric cancer in hereditary non-polyposis colorectal cancer syndrome. Int J Cancer. 1997;74:551 - 5. [PubMed: 9355980]
- Aarnio M, Sankila R, Pukkala E, Salovaara R, Aaltonen LA, de la Chapelle A, Peltomaki P, Mecklin JP, Jarvinen HJ. Cancer risk in mutation carriers of DNA-mismatch-repair genes. Int J Cancer. 1999;81:214 - 8. [PubMed: 10188721]
- Abdel-Rahman WM, Ollikainen M, Kariola R, Jarvinen HJ, Mecklin JP, Nystrom-Lahti M, Knuutila S, Peltomaki P. Comprehensive characterization of HNPCC-related colorectal cancers reveals striking molecular features in families with no germline mismatch repair gene mutations. Oncogene. 2005;24:1542 - 51. [PubMed: 15674332]
- American Cancer Society. Surveillance and health services research. Available online. 2002. Accessed 10-5-15.
- Backes FJ, Leon ME, Ivanov I, Suarez A, Frankel WL, Hampel H, Fowler JM, Copeland LJ, O'Malley DM, Cohn DE. Prospective evaluation of DNA mismatch repair protein expression in primary endometrial cancer. Gynecol Oncol. 2009;114:486 - 90. [PubMed: 19515405]
- Baglietto L, Lindor NM, Dowty DM, Wagner A, Gomez Garcia EB, Vriends AH., Dutch Lynch Syndrome Study Group. Cartwright NR, Barnetson RA, Farrington SM, Tenesa A, Hampel H, Buchanan D, Arnold S, Young J, Walsh MD, Jass J, Macrae F, Antill Y, Winship IM, Giles GG, Goldblatt J, Parry S, Suthers G, Leggett B, Butz M, Aronson M, Poynter JN, Baron JA, Le Marchand L, Haile R, Gallinger S, Hopper JL, Potter J, de la Chapelle A, Vasen HF, Dunlop MG, Thibodeau SN, Jenkins MA. Risks of Lynch syndrome cancers for MSH6 mutation carriers. J Natl Cancer Inst. 2010;102:193 - 201. [PMC free article: PMC2815724] [PubMed: 20028993]
- Bakry D, Aronson M, Durno C, Rimawi H, Farah R, Alharbi QK, Alharbi M, Shamvil A, Ben-Shachar S, Mistry M, Constantini S, Dvir R, Qaddoumi I, Gallinger S, Lerner-Ellis J, Pollett A, Stephens D, Kelies S, Chao E, Malkin D, Bouffet E, Hawkins C, Tabori U. Genetic and clinical determinants of constitutional mismatch repair deficiency syndrome: report from the constitutional mismatch repair deficiency consortium. Eur J Cancer. 2014;50:987 - 96. [PubMed: 24440087]
- Bapat B, Lindor NM, Baron J, Siegmund K, Li L, Zheng Y, Haile R, Gallinger S, Jass JR, Young JP, Cotterchio M, Jenkins M, Grove J, Casey G, Thibodeau SN, Bishop DT, Hopper JL, Ahnen D, Newcomb PA, Le Marchand L, Potter JD, Seminara D. Colon Cancer Family Registry. The association of tumor microsatellite instability phenotype with family history of colorectal cancer. Cancer Epidemiol Biomarkers Prev. 2009;18:967 - 75. [PMC free article: PMC2763617] [PubMed: 19258475]
- Barnetson RA, Tenesa A, Farrington SM, Nicholl ID, Cetnarskyj R, Porteous ME, Campbell H, Dunlop MG. Identification and survival of carriers of mutations in DNA mismatch-repair genes in colon cancer. N Engl J Med. 2006;354:2751 - 63. [PubMed: 16807412]
- Barrow E, Robinson L, Alduaij W, Shenton A, Clancy T, Lalloo F, Hill J, Evans DG. Cumulative lifetime incidence of extracolonic cancers in Lynch syndrome: a report of 121 families with proven mutations. Clin Genet. 2009;75:141 - 9. [PubMed: 19215248]
- Beamer LC, Grant ML, Espenschied CR, Blazer KR, Hampel HL, Weitzel JN, MacDonald DJ. Reflex immunohistochemistry and microsatellite instability testing of colorectal tumors for Lynch syndrome among US cancer programs and follow-up of abnormal results. J Clin Oncol. 2012;30:1058 - 63. [PMC free article: PMC3341150] [PubMed: 22355048]
- Bellizzi AM, Frankel WL. Colorectal cancer due to deficiency in DNA mismatch repair function: a review. Adv Anat Pathol. 2009;16:405 - 17. [PubMed: 19851131]
- Berends MJ, Wu Y, Sijmons RH, Mensink RG, van der Sluis T, Hordijk-Hos JM, de Vries EG, Hollema H, Karrenbeld A, Buys CH, van der Zee AG, Hofstra RM, Kleibeuker JH. Molecular and clinical characteristics of MSH6 variants: an analysis of 25 index carriers of a germline variant. Am J Hum Genet. 2002;70:26 - 37. [PMC free article: PMC384896] [PubMed: 11709755]
- Bisgaard ML, Bernstein I. HNPCC mutation rate (published abstract). Familial Cancer. 2003;2:56.
- Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA, Fodde R, Ranzani GN, Srivastava S. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58:5248 - 57. [PubMed: 9823339]
- Bouzourene H, Hutter P, Losi L, Martin P, Benhattar J. Selection of patients with germline MLH1 mutated Lynch syndrome by determination of MLH1 methylation and BRAF mutation. Fam Cancer. 2010;9:167 - 72. [PubMed: 19949877]
- Brain K, Sivell S, Bennert K, Howell L, France L, Jordan S, Rogers MT, Gray J, Sampson J. An exploratory comparison of genetic counselling protocols for HNPCC predictive testing. Clin Genet. 2005;68:255 - 61. [PubMed: 16098015]
- Burn J, Bishop DT, Mecklin JP, Macrae F, Möslein G, Olschwang S, Bisgaard ML, Ramesar R, Eccles D, Maher ER, Bertario L, Jarvinen HJ, Lindblom A, Evans DG, Lubinski J, Morrison PJ, Ho JW, Vasen HF, Side L, Thomas HJ, Scott RJ, Dunlop M, Barker G, Elliott F, Jass JR, Fodde R, Lynch HT, Mathers JC. CAPP2 Investigators. Effect of aspirin or resistant starch on colorectal neoplasia in the Lynch syndrome. N Engl J Med. 2008;359:2567 - 78. [PubMed: 19073976]
- Burt RW. Diagnosing Lynch syndrome: more light at the end of the tunnel. Cancer Prev Res (Phila) 2012;5:507 - 10. [PubMed: 22491516]
- Capelle LG, Van Grieken NC, Lingsma HF, Steyerberg EW, Klokman WJ, Bruno MJ, Vasen HF, Kuipers EJ. Risk and epidemiological time trends of gastric cancer in Lynch syndrome carriers in the Netherlands. Gastroenterology. 2010;138:487 - 92. [PubMed: 19900449]
- Chadwick RB, Pyatt RE, Niemann TH, Richards SK, Johnson CK, Stevens MW, Meek JE, Hampel H, Prior TW, de la Chapelle A. Hereditary and somatic DNA mismatch repair gene mutations in sporadic endometrial carcinoma. J Med Genet. 2001;38:461 - 6. [PMC free article: PMC1757178] [PubMed: 11474654]
- Charbonnier F, Raux G, Wang Q, Drouot N, Cordier F, Limacher JM, Saurin JC, Puisieux A, Olschwang S, Frebourg T. Detection of exon deletions and duplications of the mismatch repair genes in hereditary nonpolyposis colorectal cancer families using multiplex polymerase chain reaction of short fluorescent fragments. Cancer Res. 2000;60:2760 - 3. [PubMed: 10850409]
- Chen S, Wang W, Lee S, Nafa K, Lee J, Romans K, Watson P, Gruber SB, Euhus D, Kinzler KW, Jass J, Gallinger S, Lindor NM, Casey G, Ellis N, Giardiello FM, Offit K, Parmigiani G. Colon Cancer Family Registry. Prediction of germline mutations and cancer risk in the Lynch syndrome. JAMA. 2006;296:1479 - 87. [PMC free article: PMC2538673] [PubMed: 17003396]
- Church J, Simmang C. Practice parameters for the treatment of patients with dominantly inherited colorectal cancer (familial adenomatous polyposis and hereditary nonpolyposis colorectal cancer). Dis Colon Rectum. 2003;46:1001 - 12. [PubMed: 12907889]
- Cunningham JM, Kim CY, Christensen ER, Tester DJ, Parc Y, Burgart LJ, Halling KC, McDonnell SK, Schaid DJ, Walsh Vockley C, Kubly V, Nelson H, Michels VV, Thibodeau SN. The frequency of hereditary defective mismatch repair in a prospective series of unselected colorectal carcinomas. Am J Hum Genet. 2001;69:780 - 90. [PMC free article: PMC1226064] [PubMed: 11524701]
- de Leeuw WJ, Dierssen J, Vasen HF, Wijnen JT, Kenter GG, Meijers-Heijboer H, Brocker-Vriends A, Stormorken A, Moller P, Menko F, Cornelisse CJ, Morreau H. Prediction of a mismatch repair gene defect by microsatellite instability and immunohistochemical analysis in endometrial tumours from HNPCC patients. J Pathol. 2000;192:328 - 35. [PubMed: 11054716]
- den Bakker MA, Seynaeve C, Kliffen M, Dinjens WN. Microsatellite instability in a pleomorphic rhabdomyosarcoma in a patient with hereditary non-polyposis colorectal cancer. Histopathology. 2003;43:297 - 9. [PubMed: 12940783]
- Dinh TA, Rosner BI, Atwood JC, Boland CR, Syngal S, Vasen HF, Gruber SB, Burt RW. Health benefits and cost-effectiveness of primary genetic screening for Lynch syndrome in the general population. Cancer Prev Res (Phila) 2011;4:9 - 22. [PMC free article: PMC3793254] [PubMed: 21088223]
- Dove-Edwin I, Boks D, Goff S, Kenter GG, Carpenter R, Vasen HF, Thomas HJ. The outcome of endometrial carcinoma surveillance by ultrasound scan in women at risk of hereditary nonpolyposis colorectal carcinoma and familial colorectal carcinoma. Cancer. 2002;94:1708 - 12. [PubMed: 11920532]
- Dudok deWit AC, Tibben A, Duivenvoorden HJ, Niermeijer MF, Passchier J. Predicting adaptation to presymptomatic DNA testing for late onset disorders: who will experience distress? Rotterdam Leiden Genetics Workgroup. J Med Genet. 1998 Sep;35(9):745 - 54. [PMC free article: PMC1051427] [PubMed: 9733033]
- Duraturo F, Liccardo R, Cavallo A, De Rosa M, Grosso M, Izzo P. Association of low-risk MSH2 and MSH2 variant alleles with Lynch syndrome: probability of synergistic effects. Int J Cancer. 2011;129:1643 - 50. [PubMed: 21128252]
- Durno CA, Holter S, Sherman PM, Gallinger S. The gastrointestinal phenotype of germline biallelic mismatch repair gene mutations. Am J Gastroenterol. 2010;105:2449 - 56. [PubMed: 20531397]
- EGAPP. Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Working Group1. Recommendations from the EGAPP Working Group: genetic testing strategies in newly diagnosed individuals with colorectal cancer aimed at reducing morbidity and mortality from Lynch syndrome in relatives. Genet Med. 2009;11:35 - 41. [PMC free article: PMC2743612] [PubMed: 19125126]
- Engel C, Rahner N, Schulmann K, Holinski-Feder E, Goecke TO, Schackert HK, Kloor M, Steinke V, Vogelsang H, Möslein G, Görgens H, Dechant S, von Knebel Doeberitz M, Rüschoff J, Friedrichs N, Büttner R, Loeffler M, Propping P, Schmiegel W., German HNPCC Consortium. Efficacy of annual colonoscopic surveillance in individuals with hereditary nonpolyposis colorectal cancer. Clin Gastroenterol Hepatol. 2010;8:174 - 82. [PubMed: 19835992]
- Entius MM, Keller JJ, Drillenburg P, Kuypers KC, Giardiello FM, Offerhaus GJ. Microsatellite instability and expression of hMLH-1 and hMSH-2 in sebaceous gland carcinomas as markers for Muir-Torre syndrome. Clin Cancer Res. 2000;6:1784 - 9. [PubMed: 10815898]
- Evans DG, Gaarenstroom KN, Stirling D, Shenton A, Maehle L, Dørum A, Steel M, Lalloo F, Apold J, Porteous ME, Vasen HF, van Asperen CJ, Moller P. Screening for familial ovarian cancer: poor survival of BRCA1/2 related cancers. J Med Genet. 2009;46:593 - 7. [PubMed: 18413372]
- Fishel R, Lescoe MK, Rao MR, Copeland NG, Jenkins NA, Garber J, Kane M, Kolodner R. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell. 1993;75:1027 - 38. [PubMed: 8252616]
- Gargiulo S, Torrini M, Ollila S, Nasti S, Pastorino L, Cusano R, Bonelli L, Battistuzzi L, Mastracci L, Bruno W, Savarino V, Sciallero S, Borgonovo G, Nyström M, Bianchi-Scarrà G, Mareni C, Ghiorzo P. Germline MLH1 and MSH2 mutations in Italian pancreatic cancer patients with suspected Lynch syndrome. Fam Cancer. 2009;8:547 - 53. [PubMed: 19728162]
- Goel A, Nguyen TP, Leung HC, Nagasaka T, Rhees J, Hotchkiss E, Arnold M, Banerji P, Koi M, Kwok CT, Packham D, Lipton L, Boland CR, Ward RL, Hitchins MP. De novo constitutional MLH1 epimutations confer early-onset colorectal cancer in two new sporadic Lynch syndrome cases, with derivation of the epimutation on the paternal allele in one. Int J Cancer. 2011;128:869 - 78. [PMC free article: PMC3794437] [PubMed: 20473912]
- Gritz ER, Vernon WE, Peterson SK, Baile WF, Marani SK, Amos CI, Frazier ML, Lynch PM. Distress in the cancer patient and its association with genetic testing and counseling for hereditary non-polyposis colon cancer. Cancer Res Ther Ctrl. 1999;8:35 - 49.
- Gruber SB, Kohlmann W. The genetics of hereditary non-polyposis colorectal cancer. J Natl Compr Canc Netw. 2003;1:137 - 44. [PubMed: 19764157]
- Guilford PJ, Hopkins JB, Grady WM, Markowitz SD, Willis J, Lynch H, Rajput A, Wiesner GL, Lindor NM, Burgart LJ, Toro TT, Lee D, Limacher JM, Shaw DW, Findlay MP, Reeve AE. E-cadherin germline mutations define an inherited cancer syndrome dominated by diffuse gastric cancer. Hum Mutat. 1999;14:249 - 55. [PubMed: 10477433]
- Hamilton SR, Liu B, Parsons RE, Papadopoulos N, Jen J, Powell SM, Krush AJ, Berk T, Cohen Z, Tetu B, Burger PC, Wood PA, Taqi F, Booker SV, Petersen GM, Offerhaus GJA, Tersmette AC, Giardiello FM, Vogelstein V, Kinzler KW. The molecular basis of Turcot's syndrome. N Engl J Med. 1995;332:839 - 47. [PubMed: 7661930]
- Hampel H, Frankel W, Panescu J, Lockman J, Sotamaa K, Fix D, Comeras I, La Jeunesse J, Nakagawa H, Westman JA, Prior TW, Clendenning M, Penzone P, Lombardi J, Dunn P, Cohn DE, Copeland L, Eaton L, Fowler J, Lewandowski G, Vaccarello L, Bell J, Reid G, de la Chapelle A. Screening for Lynch syndrome (hereditary nonpolyposis colorectal cancer) among endometrial cancer patients. Cancer Res. 2006;66:7810 - 7. [PubMed: 16885385]
- Hampel H, Stephens JA, Pukkala E, Sankila R, Aaltonen LA, Mecklin JP, de la Chapelle A. Cancer risk in hereditary nonpolyposis colorectal cancer syndrome: later age of onset. Gastroenterology. 2005;129:415 - 21. [PubMed: 16083698]
- Haraldsdottir S, Hampel H, Wei L, Wu C, Frankel W, Bekaii-Saab T, de la Chapelle A, Goldberg RM. Prostate cancer incidence in males with Lynch syndrome. Genet Med. 2014;16:553 - 7. [PMC free article: PMC4289599] [PubMed: 24434690]
- Heald B, Plesec T, Liu X, Pai R, Patil D, Moline J, Sharp RR, Burke CA, Kalady MF, Church J, Eng C. Implementation of universal microsatellite instability and immunohistochemistry screening for diagnosing lynch syndrome in a large academic medical center. J Clin Oncol. 2013;31:1336 - 40. [PMC free article: PMC4878100] [PubMed: 23401454]
- Hegde M, Ferber M, Mao R, Samowitz W, Ganguly A., Working Group of the American College of Medical Genetics and Genomics (ACMG) Laboratory Quality Assurance Committee. ACMG technical standards and guidelines for genetic testing for inherited colorectal cancer (Lynch syndrome, familial adenomatous polyposis, and MYH-associated polyposis). Genet Med. 2014;16:101 - 16. [PubMed: 24310308]
- Hendriks YM, Jagmohan-Changur S, van der Klift HM, Morreau H, van Puijenbroek M, Tops C, van Os T, Wagner A, Ausems MG, Gomez E, Breuning MH, Brocker-Vriends AH, Vasen HF, Wijnen JT. Heterozygous mutations in PMS2 cause hereditary nonpolyposis colorectal carcinoma (Lynch syndrome). Gastroenterology. 2006;130:312 - 22. [PubMed: 16472587]
- Huang SC, Lavine JE, Boland PS, Newbury RO, Kolodner R, Pham TT, Arnold CN, Boland CR, Carethers JM. Germline characterization of early-aged onset of hereditary non- polyposis colorectal cancer. J Pediatr. 2001;138:629 - 35. [PubMed: 11343035]
- Iino H, Simms L, Young J, Arnold J, Winship IM, Webb SI, Furlong KL, Leggett B, Jass JR. DNA microsatellite instability and mismatch repair protein loss in adenomas presenting in hereditary non-polyposis colorectal cancer. Gut. 2000;47:37 - 42. [PMC free article: PMC1727981] [PubMed: 10861262]
- Järvinen HJ, Renkonen-Sinisalo L, Aktán-Collán K, Peltomäki P, Aaltonen LA, Mecklin JP. Ten years after mutation testing for Lynch syndrome: cancer incidence and outcome in mutation-positive and mutation negative family members. J Clin Oncol. 2009;27:4793 - 7. [PubMed: 19720893]
- Jo WS, Bandipalliam P, Shannon KM, Niendorf KB, Chan-Smutko G, Hur C, Syngal S, Chung DC. Correlation of polyp number and family history of colon cancer with germline MYH mutations. Clin Gastroenterol Hepatol. 2005;3:1022 - 8. [PubMed: 16234049]
- Kalady MF, McGannon E, Vogel JD, Manilich E, Fazio VW, Church JM. Risk of colorectal adenoma and carcinoma after colectomy for colorectal cancer in patients meeting Amsterdam criteria. Ann Surg. 2010;252:507 - 11. [PubMed: 20739851]
- Kastrinos F, Mukherjee B, Tayob N, Wang F, Sparr J, Raymond VM, Bandipalliam P, Stoffel EM, Gruber SB, Syngal S. Risk of pancreatic cancer in families with Lynch syndrome. JAMA. 2009;302:1790 - 5. [PMC free article: PMC4091624] [PubMed: 19861671]
- Kastrinos F, Steyerberg EW, Mercado R, Balmana J, Holter S, Gallinger S, Siegnund KD, Church JM, Jenkins MA, Lindor NM, Thibodeau SN, Burbidge LA, Wenstrup RJ, Syngal S. The PREMM(1,2,6) Model predicts risk of MLH1, MSH2, and MSH6 germline mutations. Gastroenterology. 2011;140:73 - 81. [PMC free article: PMC3125673] [PubMed: 20727894]
- Kawaguchi M, Yanokura M, Banno K, Kobayashi Y, Kuwabara Y, Kobayashi M, Nomura H, Hirasawa A, Susumu N, Aoki D. Analysis of a correlation between the BRAF V600E mutation and abnormal DNA mismatch repair in patients with sporadic endometrial cancer. Int J Oncol. 2009;34:1541 - 7. [PubMed: 19424571]
- Kempers MJ, Kuiper RP, Ockeloen CW, Chappuis PO, Hutter P, Rahner N, Schackert HK, Steinke V, Holinski-Feder E, Morak M, Kloor M, Büttner R, Verwiel ET, van Krieken JH, Nagtegaal ID, Goossens M, van der Post RS, Niessen RC, Sijmons RH, Kluijt I, Hogervorst FB, Leter EM, Gille JJ, Aalfs CM, Redeker EJ, Hes FJ, Tops CM, van Nesselrooij BP, van Gijn ME, Gómez García EB, Eccles DM, Bunyan DJ, Syngal S, Stoffel EM, Culver JO, Palomares MR, Graham T, Velsher L, Papp J, Oláh E, Chan TL, Leung SY, van Kessel AG, Kiemeney LA, Hoogerbrugge N, Ligtenberg MJ. Risk of colorectal and endometrial cancers in EPCAM deletion-positive Lynch syndrome: a cohort study. Lancet Oncol. 2011;12:49 - 55. [PMC free article: PMC3670774] [PubMed: 21145788]
- Kowalski LD, Mutch DG, Herzog TJ, Rader JS, Goodfellow PJ. Mutational analysis of MLH1 and MSH2 in 25 prospectively-acquired RER+ endometrial cancers. Genes Chromosomes Cancer. 1997;18:219 - 27. [PubMed: 9071575]
- Krüger S, Silber AS, Engel C, Gorgens H, Mangold E, Pagenstecher C, Holinski-Feder E, von Knebel Doeberitz M, Moeslein G, Dietmaier W, Stemmler S, Friedl W, Ruschoff J, Schackert HK. Arg462Gln sequence variation in the prostate-cancer-susceptibility gene RNASEL and age of onset of hereditary non-polyposis colorectal cancer: a case-control study. Lancet Oncol. 2005;6:566 - 72. [PubMed: 16054567]
- Kuiper RP, Vissers LE, Venkatachalam R, Bodmer D, Hoenselaar E, Goossens M, Haufe A, Kamping E, Niessen RC, Hogervorst FB, Gille JJ, Redeker B, Tops CM, van Gijn ME, van den Ouweland AM, Rahner N, Steinke V, Kahl P, Holinski-Feder E, Morak M, Kloor M, Stemmler S, Betz B, Hutter P, Bunyan DJ, Syngal S, Culver JO, Graham T, Chan TL, Nagtegaal ID, van Krieken JH, Schackert HK, Hoogerbrugge N, van Kessel AG, Ligtenberg MJ. Recurrence and variability of germline EPCAM deletions in Lynch syndrome. Hum Mutat. 2011;32:407 - 14. [PubMed: 21309036]
- Laken SJ, Petersen GM, Gruber SB, Oddoux C, Ostrer H, Giardiello FM, Hamilton SR, Hampel H, Markowitz A, Klimstra D, Jhanwar S, Winawer S, Offit K, Luce MC, Kinzler KW, Vogelstein B. Familial colorectal cancer in Ashkenazim due to a hypermutable tract in APC. Nat Genet. 1997;17:79 - 83. [PubMed: 9288102]
- Ligtenberg MJL, Kuiper RP, Chan TL, Goossens M, Hebeda KM, Voorendt M, Lee TY, Bodmer D, Hoenselaar E, Hendriks-Cornelissen SJ, Tsui WY, Kong CK, Brunner HG, van Kessel AG, Yuen ST, van Krieken JH, Leung SY, Hoogerbrugge N. Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3’ exons of TACSTD1. Nat Genet. 2009;41:112 - 7. [PubMed: 19098912]
- Lindor NM, Rabe K, Petersen GM, Haile R, Casey G, Baron J, Gallinger S, Bapat B, Aronson M, Hopper J, Jass J, LeMarchand L, Grove J, Potter J, Newcomb P, Terdiman JP, Conrad P, Moslein G, Goldberg R, Ziogas A, Anton-Culver H, de Andrade M, Siegmund K, Thibodeau SN, Boardman LA, Seminara D. Lower cancer incidence in Amsterdam-I criteria families without mismatch repair deficiency: familial colorectal cancer type X. JAMA. 2005;293:1979 - 85. [PMC free article: PMC2933042] [PubMed: 15855431]
- Lu KH, Dinh M, Kohlmann W, Watson P, Green J, Syngal S, Bandipalliam P, Chen LM, Allen B, Conrad P, Terdiman J, Sun C, Daniels M, Burke T, Gershenson DM, Lynch H, Lynch P, Broaddus RR. Gynecologic cancer as a "sentinel cancer" for women with hereditary nonpolyposis colorectal cancer syndrome. Obstet Gynecol. 2005;105:569 - 74. [PubMed: 15738026]
- Lu SL, Kawabata M, Imamura T, Akiyama Y, Nomizu T, Miyazono K, Yuasa Y. HNPCC associated with germline mutation in the TGF-beta type II receptor gene. Nat Genet. 1998;19:17 - 8. [PubMed: 9590282]
- Lynch HT, Smyrk T. Hereditary nonpolyposis colorectal cancer (Lynch syndrome). An updated review. Cancer. 1996;78:1149 - 67. [PubMed: 8826936]
- Lynch HT, Smyrk TC, Watson P, Lanspa SJ, Lynch JF, Lynch PM, Cavalieri RJ, Boland CR. Genetics, natural history, tumor spectrum, and pathology of hereditary nonpolyposis colorectal cancer: an updated review. Gastroenterology. 1993;104:1535 - 49. [PubMed: 8482467]
- Lynch HT, Watson P, Kriegler M, Lynch JF, Lanspa SJ, Marcus J, Smyrk T, Fitzgibbons RJ Jr, Cristofaro G. Differential diagnosis of hereditary nonpolyposis colorectal cancer (Lynch syndrome I and Lynch syndrome II). Dis Colon Rectum. 1988;31:372 - 7. [PubMed: 3366037]
- Machin P, Catasus L, Pons C, Munoz J, Conde-Zurita JM, Balmana J, Barnadas M, Marti RM, Prat J, Matias-Guiu X. Microsatellite instability and immunostaining for MSH-2 and MLH-1 in cutaneous and internal tumors from patients with the Muir-Torre syndrome. J Cutan Pathol. 2002;29:415 - 20. [PubMed: 12139636]
- Mathers JC, Movahedi M, Macrae F, Mecklin JP, Moeslein G, Olschwang S, Eccles D, Evans G, Maher ER, Bertario L, Bisgaard ML, Dunlop M, Ho JW, Hodgson S, Lindblom A, Lubinski J, Morrison PJ, Murday V, Ramesar R, Side L, Scott RJ, Thomas HJ, Vasen H, Gerdes AM, Barker G, Crawford G, Elliott F, Pylvanainen K, Wijnen J, Fodde R, Lynch H, Bishop DT, Burn J. CAPP2 Investigators. Long-term effect of resistant starch on cancer risk in carriers of hereditary colorectal cancer: an analysis from the CAPP2 randomised controlled trial. Lancet Oncol. 2012;13:1242 - 9. [PubMed: 23140761]
- Maxwell GL, Risinger JI, Alvarez AA, Barrett JC, Berchuck A. Favorable survival associated with microsatellite instability in endometrioid endometrial cancers. Obstet Gynecol. 2001;97:417 - 22. [PubMed: 11239648]
- Misago N, Narisawa Y. Sebaceous neoplasms in Muir-Torre syndrome. Am J Dermatopathol. 2000;22:155 - 61. [PubMed: 10770437]
- Miyaki M, Konishi M, Tanaka K, Kikuchi-Yanoshita R, Muraoka M, Yasuno M, Igari T, Koike M, Chiba M, Mori T. Germline mutation of MSH6 as the cause of hereditary nonpolyposis colorectal cancer. Nat Genet. 1997;17:271 - 2. [PubMed: 9354786]
- Moline J, Mahdi H, Yang B, Biscotti C, Roma AA, Heald B, Rose PG, Michener C, Eng C. Implementation of tumor testing for lynch syndrome in endometrial cancers at a large academic medical center. Gynecol Oncol. 2013;130:121 - 6. [PubMed: 23612316]
- Mueller-Koch Y, Vogelsang H, Kopp R, Lohse P, Keller G, Aust D, Muders M, Gross M, Daum J, Schiemann U, Grabowski M, Scholz M, Kerker B, Becker I, Henke G, Holinski-Feder E. Hereditary non-polyposis colorectal cancer: clinical and molecular evidence for a new entity of hereditary colorectal cancer. Gut. 2005;54:1733 - 40. [PMC free article: PMC1774771] [PubMed: 15955785]
- Nagasaka T, Rhees J, Kloor M, Gebert J, Naomoto Y, Boland CR, Goel A. Somatic hypermethylation of MSH2 is a frequent event in Lynch Syndrome colorectal cancers. Cancer Res. 2010;70:3098 - 108. [PMC free article: PMC2856102] [PubMed: 20388775]
- NCCN. National Comprehensive Cancer Network Guidelines for colorectal cancer screening. Available online. Registration required. 2013.
- Nicolaides NC, Carter KC, Shell BK, Papadopoulos N, Vogelstein B, Kinzler KW. Genomic organization of the human PMS2 gene family. Genomics. 1995;30:195 - 206. [PubMed: 8586419]
- Niessen RC, Hofstra RM, Westers H, Ligtenberg MJ, Kooi K, Jager PO, de Groote ML, Dijkhuizen T, Olderode-Berends MJ, Hollema H, Kleibeuker JH, Sijmons RH. Germline hypermethylation of MLH1 and EPCAM deletions are a frequent cause of Lynch syndrome. Genes Chromosomes Cancer. 2009;48:737 - 44. [PubMed: 19455606]
- Nilbert M, Therkildsen C, Nissen A, Akerman M, Bernstein I. Sarcomas associated with hereditary nonpolyposis colorectal cancer: broad anatomical and morphological spectrum. Fam Cancer. 2009;8:209 - 13. [PubMed: 19130300]
- Obermair A, Youlden DR, Young JP, Lindor NM, Baron JA, Newcomb P, Parry S, Hopper JL, Haile R, Jenkins MA. Risk of endometrial cancer for women diagnosed with HNPCC-related colorectal carcinoma. Int J Cancer. 2010;127:2678 - 84. [PMC free article: PMC2947566] [PubMed: 20533284]
- Ou J, Rasmussen M, Westers H, Andersen SD, Jager PO, Kooi KA, Niessen RC, Eggen BJ, Nielsen FC, Kleibeuker JH, Sijmons RH, Rasmussen LJ, Hofstra RM. Biochemical characterization of MLH3 missense mutations does not reveal an apparent role of MLH3 in Lynch syndrome. Genes Chromosomes Cancer. 2009;48:340 - 50. [PubMed: 19156873]
- Pande M, Amos CI, Osterwich DR, Chen J, Lynch PM, Broaddus R, Frazier ML. Genetic variation in genes for the xenobiotic-metabolizing enzymes CYP1A1, EPHX1, GSTM1, GSTT1, and GSTP1 and susceptibility to colorectal cancer in Lynch syndrome. Cancer Epidemiol Biomarkers Prev. 2008;17:2393 - 401. [PMC free article: PMC3028532] [PubMed: 18768509]
- Pande M, Lynch PM, Hopper JL, Jenkins MA, Gallinger S, Haile RW, LeMarchand L, Lindor NM, Campbell PT, Newcomb PA, Potter JD, Baron JA, Frazier ML, Amos CI. Smoking and colorectal cancer in Lynch syndrome: results from the Colon Cancer Family Registry and the University of Texas M.D. Anderson Cancer Center. Clin Cancer Res. 2010;16:1331 - 9. [PMC free article: PMC2822883] [PubMed: 20145170]
- Park YJ, Shin KH, Park JG. Risk of gastric cancer in hereditary nonpolyposis colorectal cancer in Korea. Clin Cancer Res. 2000;6:2994 - 8. [PubMed: 10955776]
- Peltomäki P. Role of DNA mismatch repair defects in the pathogenesis of human cancer. J Clin Oncol. 2003;21:1174 - 9. [PubMed: 12637487]
- Peltomäki P, Vasen H. Mutations associated with HNPCC predisposition -- Update of ICG-HNPCC/INSiGHT mutation database. Dis Markers. 2004;20:269 - 76. [PMC free article: PMC3839397] [PubMed: 15528792]
- Plaschke J, Engel C, Krüger S, Holinski-Feder E, Pagenstecher C, Mangold E, Moeslein G, Schulmann K, Gebert J, von Knebel Doeberitz M, Rüschoff J, Loeffler M, Schackert HK. Lower incidence of colorectal cancer and later age of disease onset in 27 families with pathogenic MSH6 germline mutations compared with families with MLH1 and MSH2 mutations: the German Hereditary Nonpolyposis Colorectal Cancer Consortium. J Clin Oncol. 2004;22:4486 - 94. [PubMed: 15483016]
- Ponti G, Losi L, Pedroni M, Lucci-Cordisco E, Di Gregorio C, Pellancani G, Seidenari S. Value of MLH1 and MSH2 mutations in the appearance of Muir-Torre syndrome phenotype in HNPCC patients presenting sebaceous gland tumors or keratoacanthomas. J Invest Dermatol. 2006;126:2302 - 7. [PubMed: 16826164]
- Porteous M, Dunckley M, Appleton S, Catt S, Dunlop M, Campbell H, Cull A. Is it acceptable to approach colorectal cancer patients at diagnosis to discuss genetic testing? A pilot study. Br J Cancer. 2003;89:1400 - 2. [PMC free article: PMC2394344] [PubMed: 14562005]
- Rabban JT, Calkins SM, Karnezis AN, Grenert JP, Blanco A, Crawford B, Chen LM. Association of Tumor Morphology With Mismatch-repair Protein Status in Older Endometrial Cancer Patients: Implications for Universal Versus Selective Screening Strategies for Lynch Syndrome. Am J Surg Pathol. 2014;38:793 - 800. [PubMed: 24503759]
- Rahner N, Steinke V, Schlegelberger B, Eisinger F, Hutter P, Olschwang S. Clinical utility gene card for: Lynch syndrome (MLH1, MSH2, MSH6, PMS2, EPCAM) - update 2012. Eur J Hum Genet. 2013;21(1) [PMC free article: PMC3533316] [PubMed: 22892529] [Cross Ref]
- Raymond VM, Everett JN, Furtado LV, Gustafson SL, Jungbluth CR, Gruber SB, Hammer GD, Stoffel EM, Greenson JK, Giordano TJ, Else T. Adrenocortical carcinoma is a lynch syndrome-associated cancer. J Clin Oncol. 2013a;31:3012 - 8. [PMC free article: PMC3739861] [PubMed: 23752102]
- Raymond VM, Mukherjee B, Wang F, Huang SC, Stoffel EM, Kastrinos F, Syngal S, Cooney KA, Gruber SB. Elevated risk of prostate cancer among men with Lynch syndrome. J Clin Oncol. 2013b;31:1713 - 8. [PMC free article: PMC3641694] [PubMed: 23530095]
- Renkonen-Sinisalo L, Sipponen P, Aarnio M, Julkunen R, Aaltonen LA, Sarna S, Jarvinen HJ, Mecklin JP. No support for endoscopic surveillance for gastric cancer in hereditary non-polyposis colorectal cancer. Scand J Gastroenterol. 2002;37:574 - 7. [PubMed: 12059060]
- Resnick K, Straughn JM Jr, Backes F, Hampel H, Matthews KS, Cohn DE. Lynch syndrome screening strategies among newly diagnosed endometrial cancer patients. Obstet Gynecol. 2009;114:530 - 6. [PubMed: 19701031]
- Rodriguez-Bigas MA, Vasen HF, Lynch HT, Watson P, Myrhoj T, Jarvinen HJ, Mecklin JP, Macrae F, St John DJ, Bertario L, Fidalgo P, Madlensky L, Rozen P. Characteristics of small bowel carcinoma in hereditary nonpolyposis colorectal carcinoma. International Collaborative Group on HNPCC. Cancer. 1998;83:240 - 4. [PubMed: 9669805]
- Ryan S, Jenkins MA, Win AK. Risk of prostate cancer in Lynch syndrome: a systematic review and meta-analysis. Cancer Epidemiol Biomarkers Prev. 2014;23:437 - 49. [PubMed: 24425144]
- Schulmann K, Brasch FE, Kunstmann E, Engel C, Pagenstecher C, Vogelsang H, Kruger S, Vogel T, Knaebel HP, Ruschoff J, Hahn SA, Knebel-Doeberitz MV, Moeslein G, Meltzer SJ, Schackert HK, Tympner C, Mangold E, Schmiegel W. HNPCC-associated small bowel cancer: clinical and molecular characteristics. Gastroenterology. 2005;128:590 - 9. [PubMed: 15765394]
- Schwartz RA, Torre DP. The Muir-Torre syndrome: a 25-year retrospect. J Am Acad Dermatol. 1995;33:90 - 104. [PubMed: 7601953]
- Senter L, Clendenning M, Sotamaa K, Hampel H, Green J, Potter JD, Lindblom A, Lagerstedt K, Thibodeau SN, Lindor NM, Young J, Winship I, Dowty JG, White DM, Hopper JL, Baglietto L, Jenkins MA, de la Chapelle A. The clinical phenotype of Lynch syndrome due to germline PMS2 mutations. Gastroenterology. 2008;135:419 - 28. [PMC free article: PMC2759321] [PubMed: 18602922]
- Shia J. Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome. Part 1: The utility of immunohistochemistry. J Mol Diagn. 2008;10:293 - 300. [PMC free article: PMC2438196] [PubMed: 18556767]
- Sieber OM, Lipton L, Crabtree M, Heinimann K, Fidalgo P, Phillips RK, Bisgaard ML, Orntoft TF, Aaltonen LA, Hodgson SV, Thomas HJ, Tomlinson IP. Multiple colorectal adenomas, classic adenomatous polyposis, and germline mutations in MYH. N Engl J Med. 2003;348:791 - 9. [PubMed: 12606733]
- Sijmons R, Hofstra R, Hollema H, Mensink R, van der Hout A, Hoekstra H, Kleibeuker J, Molenaar W, Wijnen J, Fodde R, Vasen H, Buys C. Inclusion of malignant fibrous histiocytoma in the tumour spectrum associated with hereditary non-polyposis colorectal cancer. Genes Chromosomes Cancer. 2000;29:353 - 5. [PubMed: 11066081]
- Sivagnanam M, Schaible T, Szigeti R, Byrd RH, Finegold MJ, Ranganathan S, Gopalakrishna GS, Tatevian N, Kellermayer R. Further evidence for EpCAM as the gene for congenital tufting enteropathy. Am J Med Genet. 2010;152A:222 - 4. [PubMed: 20034091]
- Sjursen W, Haukane BI, Grindedal EM, Aarset H, Stormorken A, Engebretsen LF, Jonsrud C, Bjørnevoll I, Andresen PA, Ariansen S, Lavik LA, Gilde B, Bowitz-Lothe IM, Maehle L, Møller P. Current clinical criteria for Lynch syndrome are not sensitive enough to identify MSH6 mutation carriers. J Med Genet. 2010;47:579 - 85. [PMC free article: PMC2976029] [PubMed: 20587412]
- South CD, Hampel H, Comeras I, Westman JA, Frankel WL, de la Chapelle A. The frequency of Muir-Torre syndrome among Lynch syndrome families. J Natl Cancer Inst. 2008;100:277 - 81. [PubMed: 18270343]
- Stoffel E, Mukherjee B, Raymond VM, Tayob N, Kastrinos F, Sparr J, Wang F, Bandipalliam P, Syngal S, Gruber SB. Calculation of risk of colorectal and endometrial cancer among patients with Lynch syndrome. Gastroenterology. 2009;137:1621 - 7. [PMC free article: PMC2767441] [PubMed: 19622357]
- Stoffel EM, Turgeon DK, Stockwell DH, Zhao L, Normolle DP, Tuck MK, Bresalier RS, Marcon NE, Baron JA, Ruffin MT, Brenner DE, Syngal S., Great Lakes-New England Clinical Epidemiology and Validation Center of the Early Detection Research Network. Missed adenomas during colonoscopic surveillance in individuals with Lynch Syndrome (hereditary nonpolyposis colorectal cancer). Cancer Prev Res (Phila) 2008;1:470 - 5. [PMC free article: PMC2671076] [PubMed: 19138994]
- Suzui M, Yoshimi N, Hara A, Morishita Y, Tanaka T, Mori H. Genetic alterations in a patient with Turcot's syndrome. Pathol Int. 1998;48:126 - 33. [PubMed: 9589476]
- Syngal S, Fox EA, Eng C, Kolodner RD, Garber JE. Sensitivity and specificity of clinical criteria for hereditary non-polyposis colorectal cancer associated mutations in MSH2 and MLH1. J Med Genet. 2000;37:641 - 5. [PMC free article: PMC1734690] [PubMed: 10978352]
- Thompson E, Meldrum CJ, Crooks R, McPhillips M, Thomas L, Spigelman AD, Scott RJ. Hereditary non-polyposis colorectal cancer and the role of hPMS2 and hEXO1 mutations. Clin Genet. 2004;65:215 - 25. [PubMed: 14756672]
- Tsai YY, Petersen GM, Booker SV, Bacon JA, Hamilton SR, Giardiello FM. Evidence against genetic anticipation in familial colorectal cancer. Genet Epidemiol. 1997;14:435 - 46. [PubMed: 9271715]
- Tutlewska K, Lubinski J, Kurzawski G. Germlie deletions in the EPCAM gene as a cause of Lynch syndrome. Hered Cancer Clin Pract. 2013;11:9. [PMC free article: PMC3765447] [PubMed: 23938213]
- Umar A, Boland CR, Terdiman JP, Syngal S, de la Chapelle A, Ruschoff J, Fishel R, Lindor NM, Burgart LJ, Hamelin R, Hamilton SR, Hiatt RA, Jass J, Lindblom A, Lynch HT, Peltomaki P, Ramsey SD, Rodriguez-Bigas MA, Vasen HF, Hawk ET, Barrett JC, Freedman AN, Srivastava S. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004;96:261 - 8. [PMC free article: PMC2933058] [PubMed: 14970275]
- van der Klift H, Wijnen J, Wagner A, Verkuilen P, Tops C, Otway R, Kohonen-Corish M, Vasen H, Oliani C, Barana D, Moller P, Delozier-Blanchet C, Hutter P, Foulkes W, Lynch H, Burn J, Moslein G, Fodde R. Molecular characterization of the spectrum of genomic deletions in the mismatch repair genes MSH2, MLH1, MSH6, and PMS2 responsible for hereditary nonpolyposis colorectal cancer (HNPCC). Genes Chromosomes Cancer. 2005;44:123 - 38. [PubMed: 15942939]
- van der Post RS, Kiemeny LA, Ligtenberg MJ, Witjes JA, Hulsbergen-van de Kaa CA, Bodmer D, Schaap L, Kets CM, van Krieken JH, Hoogerbrugge N. Risk of urothelial bladder cancer in Lynch syndrome is increased, in particular among MSH2 mutation carriers. J Med Genet. 2010;47:464 - 70. [PMC free article: PMC2991077] [PubMed: 20591884]
- Vasen HF, Mecklin JP, Khan PM, Lynch HT. The International Collaborative Group on hereditary non-polyposis colorectal cancer (ICG-HNPCC). Dis Colon Rectum. 1991;34:424 - 5. [PubMed: 2022152]
- Vasen HF, Stormorken A, Menko FH, Nagengast FM, Kleibeuker JH, Griffioen G, Taal BG, Moller P, Wijnen JT. MSH2 mutation carriers are at higher risk fo cancer than MLH1 mutation carriers: a study of hereditary nonpolyposis colorectal cancer families. J Clin Oncol. 2001;19:4074 - 80. [PubMed: 11600610]
- Vasen HF, Watson P, Mecklin JP, Jass JR, Green JS, Nomizu T, Müller H, Lynch HT. The epidemiology of endometrial cancer in hereditary nonpolyposis colorectal cancer. Anticancer Res. 1994;14:1675 - 8. [PubMed: 7979205]
- Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology. 1999;116:1453 - 6. [PubMed: 10348829]
- Vaughn CP, Hart KJ, Samowitz WS, Swensen JJ. Avoidance of pseudogene interfence in the detection of 3’ deletions in PMS2. Hum Mutat. 2011;32:1063 - 71. [PubMed: 21618646]
- Vernon SW, Gritz ER, Peterson SK, Amos CI, Perz CA, Baile WF, Lynch PM. Correlates of psychologic distress in colorectal cancer patients undergoing genetic testing for hereditary colon cancer. Health Psychol. 1997;16:73 - 86. [PubMed: 9028817]
- Wagner A, Barrows A, Wijnen JT, van der Klift H, Franken PF, Verkuijlen P, Nakagawa H, Geugien M, Jaghmohan-Changur S, Breukel C, Meijers-Heijboer H, Morreau H, van Puijenbroek M, Burn J, Coronel S, Kinarski Y, Okimoto R, Watson P, Lynch JF, de la Chapelle A, Lynch HT, Fodde R. Molecular analysis of hereditary nonpolyposis colorectal cancer in the United States: high mutation detection rate among clinically selected families and characterization of an American founder genomic deletion of the MSH2 gene. Am J Hum Genet. 2003;72:1088 - 100. [PMC free article: PMC1180263] [PubMed: 12658575]
- Wahlberg SS, Schmeits J, Thomas G, Loda M, Garber J, Syngal S, Kolodner RD, Fox E. Evaluation of microsatellite instability and immunohistochemistry for the prediction of germ-line MSH2 and MLH1 mutations in hereditary nonpolyposis colon cancer families. Cancer Res. 2002;62:3485 - 92. [PubMed: 12067992]
- Walsh MD, Buchanan DD, Cummings MC, Pearson SA, Arnold ST, Clendenning M, Walters R, McKeone DM, Spurdle AB, Hopper JL, Jenkins MA, Phillips KD, Suthers GK, George J, Goldblatt J, Muir A, Tucker K, Pelzer E, Gattas MR, Woodall S, Parry S, Macrae FA, Haile RW, Baron JA, Potter JD, Le Marchand L, Bapat B, Thibodeau SN, Lindor NM, McGuckin MA, Young JP. Lynch syndrome-associated breast cancers: clinicopathologic characteristics of a case series from the colon cancer family registry. Clin Cancer Res. 2010;16:2214 - 24. [PMC free article: PMC2848890] [PubMed: 20215533]
- Walsh MD, Buchanan DD, Pearson SA, Clendenning M, Jenkins MA, Win AK, Walters RJ, Spring KJ, Nagler B, Pavluk E, Arnold ST, Goldblatt J, George J, Suthers GK, Phillips K, Hopper JL, Jass JR, Baron JA, Ahnen DJ, Thibodeau SN, Lindor N, Parry S, Walker NI, Rosty C, Young JP. Immunohistochemical testing of conventional adenomas for loss of expression of mismatch repair proteins in Lynch syndrome mutation carriers: a case series from the Australasian site of the colon cancer family registry. Mod Pathol. 2012;25:722 - 30. [PMC free article: PMC3477239] [PubMed: 22322191]
- Watson P, Ashwathnarayan R, Lynch HT, Roy HK. Tobacco use and increased colorectal cancer risk in patients with hereditary nonpolyposis colorectal cancer (Lynch syndrome). Arch Intern Med. 2004;164:2429 - 31. [PubMed: 15596632]
- Watson P, Butzow R, Lynch HT, Mecklin JP, Jarvinen HJ, Vasen HF, Madlensky L, Fidalgo P, Bernstein I. The clinical features of ovarian cancer in hereditary nonpolyposis colorectal cancer. Gynecol Oncol. 2001;82:223 - 8. [PubMed: 11531271]
- Watson P, Lin KM, Rodriguez-Bigas MA, Smyrk T, Lemon S, Shashidharan M, Franklin B, Karr B, Thorson A, Lynch HT. Colorectal carcinoma survival among hereditary nonpolyposis colorectal carcinoma family members. Cancer. 1998;83:259 - 66. [PubMed: 9669808]
- Watson P, Vasen HF, Mecklin JP, Bernstein I, Aarnio M, Järvinen HJ, Myrhøj T, Sunde L, Wijnen JT, Lynch HT. The risk of extra-colonic, extra-endometrial cancer in the Lynch syndrome. Int J Cancer. 2008;123:444 - 9. [PMC free article: PMC2627772] [PubMed: 18398828]
- Watson P, Vasen HF, Mecklin JP, Jarvinen H, Lynch HT. The risk of endometrial cancer in hereditary nonpolyposis colorectal cancer. Am J Med. 1994;96:516 - 20. [PubMed: 8017449]
- Weissman SM, Bellcross C, Bittner CC, Freivogel ME, Haidle JL, Kaurah P, Leininger A, Palaniappan S, Steenblock K, Vu TM, Daniels MS. Genetic counseling considerations in the evaluation of families for Lynch syndrome--a review. J Genet Couns. 2011;20:5 - 19. [PubMed: 20931355]
- Westphalen AA, Russell AM, Buser M, Berthod CR, Hutter P, Plasilova M, Mueller H, Heinimann K. Evidence for genetic anticipation in hereditary non-polyposis colorectal cancer. Hum Genet. 2005;116:461 - 5. [PubMed: 15772852]
- Wijnen J, van der Klift H, Vasen H, Khan PM, Menko F, Tops C, Meijers Heijboer H, Lindhout D, Moller P, Fodde R. MSH2 genomic deletions are a frequent cause of HNPCC. Nat Genet. 1998;20:326 - 8. [PubMed: 9843200]
- Wimmer K. Relationship between NF1 and constitutive mismatch repair deficiency. In Upadhyaya M, Cooper DN, eds. Neurofibromatosis Type 1. Berlin, Germany: Springer-Verlag; 2012:235-51.
- Wimmer K, Etzler J. Constitutional mismatch repair-deficiency syndrome: have we so far seen only the tip of an iceberg? Hum Genet. 2008;124:105 - 22. [PubMed: 18709565]
- Win AK, Hopper JL, Buchanan DD, Young JP, Tenesa A, Dowty JG, Giles GG, Goldblatt J, Winship I, Boussioutas A, Young GP, Parry S, Baron JA, Duggan D, Gallinger S, Newcomb PA, Haile RW, Le Marchand L, Lindor NM, Jenkins MA. Are the common genetic variants associated with colorectal cancer risk for DNA mismatch repair gene mutation carriers? Eur J Cancer. 2013;49:1578 - 87. [PMC free article: PMC3625445] [PubMed: 23434150]
- Win AK, Young JP, Lindor NM, Tucker KM, Ahnen DJ, Young GP, Buchanan DD, Clendenning M, Giles GG, Winship I, Macrae FA, Goldblatt J, Southey MC, Arnold J, Thibodeau SN, Gunawardena SR, Bapat B, Baron JA, Casey G, Gallinger S, Le Marchand L, Newcomb PA, Haile RW, Hopper JL, Jenkins MA. Colorectal and other cancer risks for carriers and noncarriers from families with a DNA mismatch repair gene mutation: a prospective cohort study. J Clin Oncol. 2012;30:958 - 64. [PMC free article: PMC3341109] [PubMed: 22331944]
- Wu Y, Berends MJ, Mensink RG, Kempinga C, Sijmons RH, van Der Zee AG, Hollema H, Kleibeuker JH, Buys CH, Hofstra RM. Association of hereditary nonpolyposis colorectal cancer-related tumors displaying low microsatellite instability with MSH6 germline mutations. Am J Hum Genet. 1999;65:1291 - 8. [PMC free article: PMC1288281] [PubMed: 10521294]
- Zecevic M, Amos CI, Gu X, Campos IM, Jones JS, Lynch PM, Rodriguez-Bigas MA, Frazier ML. IGF1 gene polymorphism and risk for hereditary nonpolyposis colorectal cancer. J Natl Cancer Inst. 2006;98:139 - 43. [PubMed: 16418517]
- Zhang L. Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome. Part II: The utility of microsatellite instability testing. J Mol Diagn. 2008;10:301 - 7. [PMC free article: PMC2438197] [PubMed: 18556776]
推荐阅读
- Hampel H, Panescu J, Lockman J, Sotamaa K, Fix D, Comeras I, LaJeunesse J, Nakagawa H, Westman JA, Prior TW, Clendenning M, de la Chapelle A, Frankel W, Penzone P, Cohn DE, Copeland L, Eaton L, Fowler J, Lombardi J, Dunn P, Bell J, Reid G, Lewandowski G, Vaccarello L. Comment on: Screening for Lynch syndrome (hereditary nonpolyposis colorectal cancer) among endometrial cancer patients. Cancer Res. 2007;67:9603. [PubMed: 17909073]
- Rumilla K, Schowalter KV, Lindo NM, Thomas BC, Mensink KA, Gallinger S, Holter S, Newcomb PA, Potter JD, Jenkins MA, Hopper JL, Long TI, Weisenberger DJ, Haile RW, Casey G, Laird PW, Le Marchand L, Thibodeau SN. Frequency of deletions of EPCAM (TACSTD1) in MSH2-associated Lynch syndrome cases. J Mol Diagn. 2011;13:93 - 9. [PMC free article: PMC3069927] [PubMed: 21227399]
本章注释
修改历史
- 2014年5月22日 (me) 全面更新发布
- 2012年9月20日 (cd) 修订:临床上可获得的Lynch综合征(遗传性非息肉病性结肠癌)多基因检测组合
- 2011年8月11日 (me) 全面更新发布
- 2006年11月29日 (me) 全面更新网络发布
- 2004年2月5日 (me) 评论网络发布
- 2003年4月18日 (sg) 原始提交