
概述
临床特征.
天使综合征(AS)的特征是严重的发育迟缓或智力障碍,明显的语言能力受损、共济失调步态和/或肢体震颤,伴有频繁大声笑、微笑和易兴奋等异常快乐表现的独特行为。常见小头畸形和癫痫发作。发育迟缓从6个月左右开始;但是,1岁之后AS的特异性临床症状才会出现,通常临床诊断需要数年时间。
诊断/检测.
临床诊断是指先证者符合临床诊断标准共识和/或分子遗传学检测结果表明母源遗传的UBE3A等位基因表达或功能缺陷。约80%的AS患者可通过15q11.2-q13染色体区域的亲本特异性DNA甲基化印记分析进行检测,包括缺失、单亲二体(UPD)或印迹缺陷(ID)三种分子缺陷。不足1%的个体是由于细胞遗传学可见的染色体重排(即易位或倒位)所致。约11%的个体经序列分析可检测到UBE3A基因的致病性变异。因此,分子遗传学检测(甲基化分析和UBE3A序列分析)能够确定大约90%患者的遗传变异。其余10%具有AS典型表型特征的个体由于其遗传机制尚未明确,因此不适用于上述诊断检测。疾病管理.
对症治疗:常规处理喂养困难,便秘,胃食管反流和斜视。抗癫痫药物用于治疗癫痫发作。给予理疗,作业疗法以及重点强调非语言沟通方式的语言治疗,包括辅助工具(如图片卡或交流板)加强沟通和书写。学校中个性化和灵活的教学方式。 镇静剂治疗夜间觉醒。矫正背心和/或手术干预治疗脊柱侧弯。
继发性并发症的预防:癫痫患儿存在药物过度治疗的风险,因为异常运动可能被误认为是癫痫发作以及惊厥控制后脑电图异常持续存在。镇静剂如利培酮或其他非典型抗精神病药物可以引起副作用。
监测:每年随访脊柱侧弯的情况。评估年长儿童因过度进食所导致的肥胖。
需要避免使用的药物:由于卡马西平,氨己烯酸和噻加宾可能会加重癫痫发作。
遗传咨询 .
AS是由于位于15q11.2-q13天使综合征/ 普拉德-威力综合征(AS/PWS)区域内母源印记的UBE3A基因失功能所致。先证者同胞的风险取决于导致UBE3A功能丧失的遗传机制:对于缺失或UPD的先证者,其同胞受累风险通常小于1%。对于存在ID或UBE3A致病性变异的先证者,其同胞受累风险通常高达50%,先证者母亲的其它家庭成员的患病风险也会增加。细胞遗传学可见的染色体重排通常是新发的,但也有可能是遗传的。当遗传机制确定为缺失,UPD,ID,UBE3A致病变异或染色体重排时,需要对具有风险的妊娠进行产前检测。
诊断
天使综合征(AS)临床诊断标准共识是由美国天使综合征基金会的科学顾问委员会共同制定的 [Williams et al 2006]。近期数篇综述可供查阅[Dagli et al 2012, Thibert et al 2013, Bird 2014].
支持性证据
以下临床和/或实验室检查结果可以支持天使综合征的诊断。
临床表现
新生儿通常具有正常的表型。 发育迟缓首先出现在六个月左右。但是,一岁后AS的特征性临床表现才会出现,通常临床诊断需要数年的时间。
受累的个体中出现的典型表现
- 胎儿期和出生史正常,出生时头围正常,没有重大出生缺陷
- 代谢、血液和生化方面的实验室检查结果均正常
- MRI或CT检查提示脑结构正常,但可观察到轻度的皮层萎缩或髓鞘形成障碍
- 发育里程碑延迟,但没有技能缺失
- 6至12个月时出现发育迟缓,最终归类为重度发育迟缓
- 言语障碍,轻者用词困难;接受语言能力和非口头沟通能力优于表达语言能力
- 运动或平衡障碍,通常是步态共济失调和/或肢体震颤
- 行为独特,包括频繁大声笑/微笑;明显的快乐举止、兴奋,往往伴有拍手和多动
超过80%受累的个体中出现的表现
- 头围小或增长缓慢,通常在两岁时导致明确的或相对的小头畸形
- 通常在三岁之前出现癫痫发作
- 异常脑电图,具有特征性高波幅棘慢波
低于80%受累的个体中出现的表现
- 平枕
- 枕部凹陷
- 吐舌
- 舌头前顶;吸吮/吞咽障碍
- 婴儿期喂养问题和/或肌张力减退
- 下颌突出
- 宽嘴,牙齿缝隙宽
- 频繁流涎
- 过度的咀嚼/嘴部动作
- 斜视
- 皮肤色素减退,发色与眼睛颜色淡(与家人相比);仅见于缺失患者
- 下肢深腱反射亢进
- 上肢抬起、弯曲,特别是在行走时更为明显
- 步基宽,足内翻或外翻
- 热敏感性增加
- 睡眠-觉醒周期异常,睡眠减少
- 迷恋水;喜欢皱纹物品如某些纸张和塑料
- 与食物有关的异常行为
- 肥胖(在年龄较大的儿童中出现;在非缺失患者中更常见)
- 脊柱侧弯
- 便秘

图 1 .所述个体均得到了天使综合征的基因确诊。通常可观察到快乐表情和步态不稳,通常伴随着上肢抬起。 有时,面部外观可以提示诊断,但通常面部特征并不特异 (更多信息请点击此处)
明确诊断
符合临床诊断标准共识和/或分子遗传学检测结果表明母源遗传的UBE3A等位基因表达或功能缺陷(参见表 1)的先证者可通过以下机制之一明确AS诊断:
基于提示AS的临床表现和实验室结果进一步进行分子遗传学检测,以明确诊断。
有临床症状但尚未进行过任何分子遗传学检测的个体:
通过染色体芯片(CMA),荧光原位杂交(FISH)或核型分析*发现的15q11.2-q13缺失个体,进行DNA甲基化分析,以确定缺失是否发生在母源15号染色体。
* 低于1%的AS患者在细胞遗传学分析可见一条涉及15q11.2-q13区段的15号 染色体重排(即易位或倒位)。
表1.
天使综合征的检测方法
| 检测方法 | 检测到的遗传机制 1 | 检测方法所发现的AS患者比例2 | ||||
|---|---|---|---|---|---|---|
| 15q11.2-q13缺失 | UPD | ID |
| UBE3A缺失/重复 | ||
| DNA 甲基化分析 3, 4 | X | X | X 5 | ~80% | ||
| MS-MLPA 6 | X | X | X | ~80% | ||
| FISH 7 | X | ~68% | ||||
| CMA 8 | X | X 9 | ~68% | |||
| UPD研究 10 | X | ~7% | ||||
| AS IC 缺失分析 11, 12 | X | ~3% | ||||
| UBE3A序列分析 | X 13 | ~11% 14 | ||||
| UBE3A基因的缺失/重复分析 11, 15 | X | 罕见 | ||||
- 1.
详细信息请见分子遗传。
- 2.
11%的临床拟诊的AS个体,在本表所述的所有检测结果都正常。
- 3.
- 4.
不能区分遗传机制
- 5.
80%-90%IDs被认为是母体卵子发生或早期胚胎发生期间出现了表观遗传致病变异[Buiting 2010]。主要通过科研实验室可将ID分为IC缺失或表观遗传缺陷。
- 6.
甲基化特异性多重连接依赖性探针扩增(MLPA)可以与甲基化分析扩增法一同用于检测缺失 [Nygren et al 2005, Procter et al 2006, Ramsden et al 2010]。
- 7.
- 8.
- 9.
- 10.
利用DNA多态性标记可检测UPD,这需要来自受累个体及其父母的DNA样品。
- 11.
- 12.
AS印记中心(IC)的缺失分析可检测小缺失(6-200kb),占所有印记缺陷(IDs)病例的8%-15%[Buiting 2010]。
- 13.
- 14.
- 15.
尽管CMA通常检测15q11.2-q13的大片段缺失,但在极少数情况下,CMA可以检测到UBE3A基因多个外显子或整个基因缺失[Lawson-Yuen et al 2006, Sato et al 2007]。
11%或更多经临床诊断但未能检测出遗传异常的AS患者,可能解释如下:
- 临床诊断错误
- 不在检测范围的UBE3A基因调控区致病性变异
- 影响UBE3A功能的其他未知机制或基因
临床特征
临床描述
胎儿期、出生体重和出生头围通常正常。天使综合征(AS)婴儿可能会有母乳或人工喂养困难(由于吸吮困难)和肌张力低下。可能会出现胃食管反流。
有些婴儿会产生明显的快乐情绪,伴有大笑或阵发笑。50%的患儿在12个月前会出现小头畸形。斜视也可能出现。12月龄前可能会观察到震颤伴深反射活跃。
因为大动作发育延迟、肌张力减退和/或语言发育延迟,AS首先可能会在幼儿期拟诊[Williams et al 2006]。
癫痫通常发生在1岁到3岁之间,可能伴发非特异性或特异性的脑电图改变:成串高波幅δ活动及阵发性棘慢波发放(有时可见切迹样δ波);广泛的成串节律性θ活动;后1/3头部成串的节律性5-6/s的尖样θ波,形成小棘波复合波。这些波形常在闭眼时出现或增强。[Boyd et al 1997, Rubin et al 1997, Korff et al 2005]。
癫痫发作类型多样,包括运动为主的发作和小运动的发作(例如小发作、失张力发作) [Galván-Manso et al 2005, Pelc et al 2008a, Thibert et al 2009, Fiumara et al 2010]。婴儿痉挛很少见。非惊厥性癫痫持续状态可能会出现 [Pelc et al 2008a]。脑部MRI显示轻度萎缩和轻度髓鞘形成,但无结构性病变 [Harting et al 2009, Castro-Gago et al 2010]。
AS患儿平均行走的年龄在2.5岁至6岁之间 [Lossie et al 2001] ,这个阶段可能会出现一种呆板的机器人样僵硬步态,伴有前臂上抬、弯曲和前倾,过度兴奋、大笑、爱伸舌、流口水、缺乏语言和寻求社交行为。10%的孩子不能行走。
AS患者的睡眠问题较普遍;夜间频繁觉醒较常见 [Bruni et al 2004, Didden et al 2004]。已报道有睡眠异常(睡眠开始或维持困难)、睡眠-觉醒周期不规则、破坏性夜间行为比如阵发性笑声和睡眠相关癫痫发作 [Pelc et al 2008b]。
基本上所有的AS患儿都会有一些多动;男性和女性似乎均受累。婴幼儿可能看似不停地活动,不断地将手或玩具放在嘴里,和/或持续更换物品。一些行为可能提示存在自闭症谱系的问题,但社会交往情况通常良好,很少出现刻板行为,例如排列玩具或迷恋旋转物体或闪烁灯 [Walz 2007]。
语言障碍严重。罕见能够恰当地使用一个或两个单词。接受语言能力通常优于表达语言能力[Gentile et al 2010]。大多数年龄较大的AS儿童和成年AS患者能够通过手势以及使用交流板进行交流。不会有效流畅地使用手语[Clayton-Smith 1993]。嵌合性印记缺陷患者的语言能力相对较高。
AS患者进入青春期的时间以及青春发育一般正常。生育能力似乎正常;男性和女性患者均可能生育。 Lossie & Driscoll [1999] 报道受累母亲将AS缺失传递给胎儿。
尽管整个成年期患者仍会出现癫痫发作,但总体而言年轻个体的身体健康状况似乎良好。便秘较为常见。许多个体会进行胃食管反流症状的治疗。随着年龄的增长,脊柱侧弯更为常见[Giroud et al 2015, Larson et al 2015]。
AS成年个体难以独立生活;许多患者会居家或在类似家的地方生活。
生存数据目前尚无,但寿命几乎正常。
基因型-表型相关性
AS所有遗传机制均会导致患者出现一致的临床特点:重度到极重度的智力残疾、运动障碍、特征行为以及严重发音和语言受限。然而,一些临床差异与基因型相关[Fridman et al 2000, Lossie et al 2001, Varela et al 2004, Tan et al 2011, Valente et al 2013]。这些相关性大致总结如下:
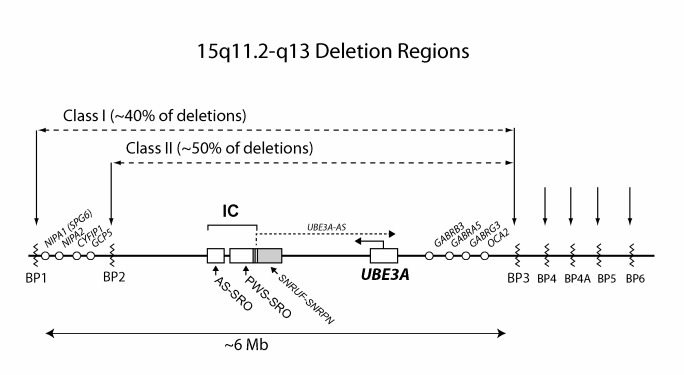
- 5-7Mb缺失的类型会导致出现最严重的表型,有小头畸形、癫痫发作、运动障碍(例如共济失调、肌张力低下、喂养困难)和语言障碍。与具有UPD或印记缺陷的个体相比,他们的体重指数也较低 [Tan et al 2011]。有一些观点认为,与BP2-BP3(II类; ISCA-37478)断点缺失型个体相比,较大缺失范围的个体(例如BP1-BP3 [I类; ISCA-37404]断点)可能存在更多的语言障碍或自闭症特征[Sahoo et al 2006] (见图2)。
- 与具有其他分子机制的患者相比,UPD个体的身体发育情况更好(较少发生小头畸形),运动异常较少、共济失调较轻、癫痫发生率较低(但并非不存在) [Lossie et al 2001, Saitoh et al 2005, Valente et al 2013].。
- 与具有其他分子机制的患者相比,IDs或UPD个体的发育情况更好、语言能力更佳。对于非缺失型ID(大约占ID组的20%)的个体,嵌合型的语言能力最好[Nazlican et al 2004];他们可以说50-60个词汇,并会使用简单的句子。
- 包含OCA2在内的染色体缺失个体通常会存在虹膜、皮肤和毛发色素减退。OCA2编码酪氨酸代谢中一种重要的蛋白质,与皮肤、毛发和虹膜中色素的发生有关(参见2型眼皮肤白化病)。然而,除了OCA2单倍剂量不足以外,其他因素似乎也可以解释AS患者色素相对不足的情况,现已显示UBE3A可调节体细胞组织中的黑皮质素1受体(MC1R)的活性 [Low & Chen 2011]。
外显率
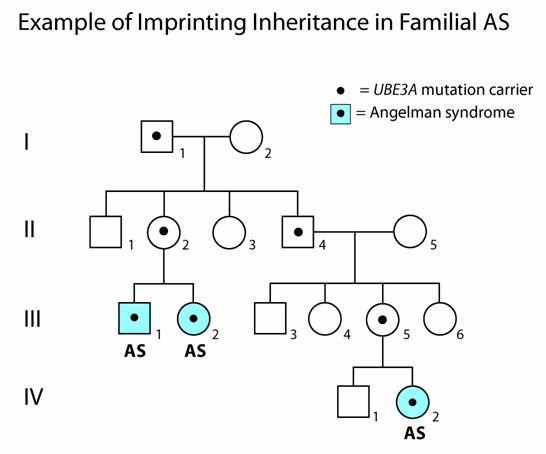
遗传性UBE3A致病性变异、IC缺失、包括UBE3A在内的较小的15q11.2-q13缺失 [Kuroda et al 2014] 和某些染色体易位,在遵循印记(或遗传)模式时,如果遗传了父源致病变异的个体则无症状发生(见图3)。
患病率
AS在人群中的患病率为1/12,000-24,000[Clayton-Smith & Pembrey 1992, Steffenburg et al 1996, Mertz et al 2013]。
遗传相关(等位基因)疾病
普拉德-威利综合征(PWS)是由于父源15q11.2-q13区域丢失所致。虽然年龄较大的PWS和AS儿童,其临床表现有所不同,但两岁以下儿童的一些临床表现会有重叠,例如;喂养困难、张力减退、发育迟缓 [Cassidy et al 2000]。
母源染色体15q11.2-q13区域的重复会引发临床上不同于AS或PWS的疾病。15q11.2-q13重复的个体并无面部畸形,但有轻度到中重度学习障碍,并且可能出现孤独症谱系行为 [Boyar et al 2001]。可参阅15q 重复综合征和相关疾病。
鉴别诊断
AS婴儿通常存在非特异性精神运动发育迟缓和/或癫痫; 因此,鉴别诊断常常具有广泛性和非特异性,包括脑瘫、静止性脑病或线粒体脑肌病。大多数AS婴儿出现的震颤和肢体痉挛动作可能有助于将AS与这些疾病进行区别。
以下与AS类似的疾病需要在鉴别诊断中考虑[Tan et al 2014]:
- Mowat-Wilson综合征 可表现为快乐情绪、癫痫发作、下颌突出、耳垂上翻、言语能力减退、小头畸形、便秘,偶尔出现先天性巨结肠[Zweier et al 2005]。先天性心脏缺陷或胼胝体发育不全也可发生。Mowat-Wilson综合征通常是ZEB2基因出现显性的新发致病性变异或该基因的缺失所致。
- Pitt-Hopkins 综合征(PTHS)的特征有智力障碍、宽嘴和特殊面容以及间歇性过度通气伴随呼吸暂停 [Zweier et al 2007]。可能与AS重叠的特征包括小头畸形、癫痫、共济失调的步态和快乐性格[Takano et al 2010]。一些患儿在3岁以后出现白天过度通气的特征 [Peippo et al 2006]。PTHS是由TCF4基因的致病性变异或该基因所在染色体 区域缺失 (18q21.2)导致的TCF4单倍体剂量不足所致。迄今为止,报道的大多数 受累个体均是由新发的致病性变异或缺失所致的散发病例(即一个家族中有单个个体发病)。
- Christianson综合征与AS也较为类似。临床特征包括明显快乐情绪、认知严重延迟、共济失调、小头畸形和癫痫发作 [Christianson et al 1999, Gilfillan et al 2008, Schroer et al 2010]。受累个体可能身材瘦弱,10岁后可能会丧失行走能力。一些患者会出现小脑和脑干萎缩[Gilfillan et al 2008]。尽管癫痫在两种疾病中都存在,但EEG表现似乎有所不同:AS典型地表现为广泛的高波幅慢/棘慢波(1.5-3 Hz),而SLC9A6致病性变异的个体缺乏AS EEG的特点,表现为背景频率更快(10-14Hz)的模式 [Gilfillan et al 2008]。Christianson综合征是由SLC9A6基因突变引起的X连锁疾病。
- 表现为癫痫、后天性小头畸形和严重语言障碍的AS女婴可能与Rett 综合征(OMIM 312750)女童相似。Rett综合征女童通常不具有明显的快乐行为,AS女童不会有发育倒退过程或双手失用。年龄较大的未被诊断为Rett综合征的女性可能具有类似于AS的特征 [Watson et al 2001]。Rett综合征是由MECP2基因突变引起的 X-连锁疾病。
- 有时,表现为喂养困难和肌张力减退的AS婴儿,经过染色体芯片或FISH检测到15q11.2-q13缺失,但没有通过DNA甲基化分析证实为母源缺失,而会被误诊为Prader-Willi综合征。
- 涉及MBD5基因的2q23.1微缺失可能会导致严重的言语延迟、癫痫、行为障碍和小头畸形。一些个体会出现AS样的表型 [van Bon et al 2010, Williams et al 2010]。可参阅MBD5单倍体剂量不足。
- 其他染色体疾病,特别是 22q13.3 缺失综合征(Phelan-McDermid综合征)[Precht et al 1998]会出现AS的一些特点。22q13.3缺失综合征的特征表现为无面部畸形、言语缺失或极少、中度至重度发育迟缓,有时会伴有自闭症谱系的行为特征。其他微缺失综合征,特别是染色体芯片检测到的新微缺失综合征,可能也与AS的某些特征有关 [Brunetti-Pierri et al 2008, Sharkey et al 2009]。
- 男性MECP2 重复(通常包含Xq28约500kb的区域)的临床特征有严重发育障碍、言语缺失、癫痫和共济失调步态伴随痉挛性截瘫。虽然成年男性患者通常不能行走,并容易出现感染性疾病,但儿童可能存在非特异性表现,包括智力残疾伴自闭症、言语缺失和不稳定步态等特征[Van Esch et al 2005, Friez et al 2006, Lugtenberg et al 2009]。 MECP2重复 综合征的遗传方式为X连锁。
- 腺苷酸琥珀酸裂解酶缺乏症 (OMIM 103050)会导致琥珀酰嘌呤蓄积,引起精神运动迟缓、自闭症、肌张力低下和癫痫发作 [Spiegel et al 2006]。 已有报告显示女性患者中存在运动性失用、严重的言语缺陷、过度发笑、快乐特质、多动、注意力短暂、嘴部咀嚼动作、发脾气和刻板动作 [Gitiaux et al 2009]。 实验诊断涉及脑脊液、尿液和(较少量)血浆中检测琥珀酰基氨基咪唑甲酰胺核糖苷(SAICA核糖苷)和琥珀酰腺苷(S-Ado)。腺苷酸琥珀酸裂解酶缺乏症的遗传方式为常染色体隐性遗传,由ADSL基因致病性变异所引起。
- 已报告的一名男性患儿患有罕见的与低甲硫氨酸和高同型半胱氨酸血症相关的代谢紊乱疾病-重度亚甲基四氢叶酸还原酶(MTHFR)缺乏症 (OMIM236250),患儿伴有快乐举止、共济失调步态、言语缺失和枕部扁平等特征 [Arn et al 1998]。MTHFR缺乏症的遗传方式为常染色体隐性遗传 ,由MTHFR基因的致病性变异所引起。
- 罕见情况下,先天性糖基化障碍(CDG)可以出现AS类似的特征,受累 儿童出现不稳定步态、言语障碍和癫痫。
- Kleefstra综合征是由染色体9q34.3区域的EHMT1基因单倍体剂量不足所致 [Willemsen et al 2012] 。 AS和Kleefstra综合征均已报告的共同的临床特征包括:中度至严重的智力障碍,语言减少;与表达性语言技能相比,接受性语言技能更好;儿童期存在肌张力减低;睡眠障碍(多次觉醒;人中短伴有下颌突出。 区分Kleefstra综合征与AS的面部特征包括连眉和下唇外翻。 一些轻度的Kleefstra综合征患者可以拥有超过100个字的词汇量,并会说成句的话,这在AS患者中非常罕见。
- HERC2相关的认知障碍 (OMIM 615516)。 HERC2位于 染色体15q13.1,编码一种能够结合并影响E6AP泛素连接酶活性的蛋白。 在4个阿米什家族和1个阿米什-门诺农混血家族的22名成员中发现了HERC2基因c.1781C> T(p.Pro594Leu) 纯合性致病性变异,患者存在全面发育迟缓、智力障碍和肌张力减低,独走延迟至2.5岁-5岁之间,跑动时步基宽,并伴随胳膊上抬和肘部弯曲 [Harlalka et al 2013]。 这些表现让人联想到那些轻度受累的AS患者,但与AS的区别在于这些患者缺乏易笑的表现,且智力障碍相对较轻。
- WAC-related intellectual disability(ID)是由WAC(参与转录调节的基因)中的杂合致病性变异引起的。大多数受累婴儿在出生时具有显著但非特异性的特征,例如新生儿张力减低和喂养问题。临床上很少怀疑WAC相关ID的诊断;通常通过Panel或全外显子组测序提供疾病诊断。AS和WAC相关智力障碍都具备的临床特征包括智力残疾,语言延迟,睡眠障碍,癫痫发作和颅面变化(例如,相对大的嘴和突出的下巴/下颌骨)。然而,与AS相比(严重智力缺陷),WAC相关智力障碍的患者通常较少个体为严重智力缺陷(轻度至重度),拥有较好的言语能力(例如,通常可以说出单词和句子);癫痫发作率较低,并且没有小头畸形[DeSanto et al 2015, Lugtenberg et al 2016]。迄今为止,已有18个人被确定为与WAC相关的智力障碍。
管理
初次诊断后进行评估
为了确定诊断为天使综合征(AS)的个体的疾病程度和需求,建议进行如下评估,重点是神经系统评估和积极预防:
对症治疗
新生儿喂养困难可能需要采用特殊奶嘴和其他策略,以便对吮吸能力弱或不协调的情况进行管理。
胃食管反流可能与体重不增和呕吐有关;传统的医疗方法(比如垂直体位、胃动力药物)通常有效;有时应根据需要进行胃底折叠术。
许多抗癫痫药物(AEDs)已被用于治疗AS患者的癫痫发作,暂无任何一种药物显示具有优势。用于治疗小运动发作的药物(如丙戊酸、氯硝西泮、托吡酯、拉莫三嗪、乙琥胺)比大运动发作药物(如苯妥英、苯巴比妥)更常用 [Thibert et al 2009]。卡马西平虽然并无禁忌症,但比其他常用抗惊厥药物的使用率低。优先选用单一药物治疗,但突发癫痫发作很常见。少数AS患者偶有癫痫发作,且未进行AEDs治疗。一些难治性癫痫患者曾受益于生酮或低血糖指数饮食 [Thibert et al 2012]。
行为治疗往往对多动无效;家庭的调整及为患儿提供安全的环境非常重要。
大多数AS患儿不会因多动接受药物治疗,但有些患者可能受益于兴奋剂药物,如哌甲酯(Ritalin®)。
行为矫正对于治疗社会破坏性或自我伤害的不良行为是有效的。
应该提供全面的教育培训和内容丰富培训项目。
行动不便或不能行走的患儿可能受益于物理治疗。职业治疗有助于改善精细运动和控制口部活动。对于严重共济失调的患儿需要特殊的矫正椅或固定器具。
语言治疗至关重要,应该关注非语言沟通方式。应在适当的时候尽早使用辅助交流工具,如图片卡或交流板。一旦孩子注意力足够集中应立即开始尝试教授书写。
学校的个性化和灵活性管理对患儿来说是重要的教育策略。
为了有效地融入课堂,可能需要在教室里配备特殊的物理设备,以及教师助手或助教。多动的AS患儿需要一间宽敞的教室。
许多家庭建造安全但有限制的卧室,以适应夜间的觉醒。睡前一小时服用0.3毫克褪黑激素可能对某些患儿有帮助。但如果孩子半夜醒来,则不应服用。
斜视可能需要手术矫正。
便秘通常需要定期使用泻药,如高纤维或润滑药物。
矫形问题,特别是半脱位、足内翻或跟腱挛缩,可通过矫形器具或手术来矫正。
脊柱侧弯需要矫形背心治疗,严重弯曲的患者可能获益于外科手术的支架。
预防继发性并发症
AS患儿存在药物过度治疗的风险。由于AS患儿的运动异常可能被误认为癫痫发作,并且即使癫痫发作得到控制,EEG异常也可能持续存在。
天使综合征的行为表型包括易兴奋、多动和社交技能缺陷。这些问题会使他们具有社交隔绝的风险。有时,使用利培酮(Risperdal®)或其他非典型抗精神病药物会有一些益处,但作用通常有限。当需要这种药物时,必须谨慎,避免出现镇静过度和其他副作用。
较年长的成人患者容易出现活动减少和兴奋性降低的情况,安排活动可能会有助于减少脊柱侧弯和肥胖的程度。
监管
以下情况是适当的:
- 每年检查脊柱侧弯
- 对于年龄较大的儿童,评估与食欲过盛和体力活动减少相关的肥胖情况。
避免的药物/环境
虽然卡马西平并无禁忌症,但比其他常用的抗惊厥药物的使用率低。
天使综合征患者禁止使用氨己烯酸和噻加宾(可增加大脑GABA水平的抗惊厥药)。尽管原因不明,但卡马西平、氨己烯酸和替加宾可引起其他癫痫发作类型或非惊厥性癫痫持续状态。这种反常性的癫痫发作并不仅仅局限在天使综合征患者[Pelc et al 2008a]。
患者亲属的风险评估
正在研究的治疗方法
口服叶酸、维生素B12、肌酸和甜菜碱的临床试验已经开展,旨在增加DNA甲基化途径,并可能增加父源UBE3A等位基因在中枢神经系统中的表达。然而,最初的试验并未显示明显的临床疗效 [Peters et al 2010](更多信息详见全文 )。最新的治疗研究主要是使用端粒酶抑制剂[Huang et al 2011] 和反义寡核苷酸[Meng et al 2015]激活沉默的父源UBE3A等位基因。搜索ClinicalTrials.gov网址,可以获取关于疾病临床研究的相关信息。
其他
过度伸舌引起流涎;常规的手术或药物治疗(例如,唾液导管的手术再植或局部使用东莨菪碱贴剂)通常无效。
遗传咨询
遗传咨询是向个人和家庭提供有关遗传疾病的性质、遗传方式和影响的信息,以帮助他们做出知情的医疗决策和个人决定的过程。下述部分涉及遗传风险评估、家族史和基因检测,阐明家庭成员的遗传状况。本部分的目的并非要解决个人可能面临的所有私人、文化或伦理问题,也并非要替代遗传学专业人员的咨询。-ED。
遗传方式
家庭成员的风险
先证者的父母
先证者的同胞 AS个体的同胞,其患病风险取决于AS先证者的遗传机制(总结在表 2中)。
再发风险的评估 如果DNA甲基化模式显示存在母源缺失的特征,则应明确潜在的遗传机制(缺失、UPD或ID),以进行遗传咨询。
表 2.
不同AS遗传机制先证者的同胞受累风险
| 分子遗传类型1 | 比例 | 遗传机制 | 同胞患病再发风险 |
|---|---|---|---|
| Ia | 65%-75% | 5-至7-Mb 缺失 | <1% |
| Ib | <1% | 染色体不平衡易位或者遗传的小片段染色体缺失 | 可能高达50% |
| IIa | 3%-7% | 父源UPD | <1% |
| IIb | <1% | 父源UPD with predisposing parental 易位 | 如果父亲携带15;15罗伯逊易位则风险接近100% |
| IIIa | 0.5% | IC缺失型 ID | 如果母亲携带IC 缺失则风险高达50% |
| IIIb | 2.5% | IC未见缺失型ID | <1% |
| IV | 11% | UBE3A致病性变异 | 如果母亲携带致病性变异则风险高达50% |
| V | 10%-15% | "其他" - 未发现已证实的分子异常 | 风险不明 |
1.基于Jiang et al [1999]所得的术语:
Ia.缺失型患者的母亲应该进行染色体核型和 FISH分析,以确定她们是否存在染色体重排。对于存在新发大片段缺失的先证者而言,同胞的风险低于1%。这些大片段缺失出现生殖嵌合的情况已有报告[Kokkonen & Leisti 2000, Sánchez et al 2014]。
Ib.如果先证者体内发现染色体重排或基因区域的小的缺失,同胞和其他家族成员的风险取决于重排是遗传或是新发 [Horsthemke et al 1996, Stalker & Williams 1998, Gimelli et al 2003, Collinson et al 2004, Kuroda et al 2014]。
IIa.在父源UPD型AS家系以及先证者体内并未鉴定到罗伯逊染色体易位的家族中,同胞患AS的风险低于1%。这个风险值是基于如下三方面考虑所得:所有已知的染色体正常的UPD病例缺乏再发情况,其他病症中的UPD的经验以及UPD机制的理论考虑。已经观察到母源15号染色体存在复发的减数分裂不分离,因此再发风险不为零 [Harpey et al 1998]。
如果AS个体是由于父源UPD所致,且染色体核型正常,则应该为其母亲进行染色体分析,以排除罕见的罗伯逊易位或标记染色体这些易感因素(例如,产生15号染色体缺失的母系配子,随后通过合子“挽救”形成父源单亲二体)。
IIIa.印记中心(IC)缺失的患者,其表型正常的母亲也有可能 IC缺失。母亲的 IC缺失可能是新发的父源15号染色体的致病性变异,也可能是遗传了其父亲的IC缺失,这与调控15q11.2-q13区域的印记机制一致 [Buiting et al 2001, Horsthemke & Buiting 2008]。
另外,其中一些母亲可能存在IC缺失的 胚系嵌合现象。当IC缺失先证者的母亲的外周血遗传研究结果正常时,遗传咨询会变得复杂。如果先证者的母亲存在已知的IC缺失,同胞的风险是50%。
IIIb. 迄今为止,在IC没有缺失的IDs先证者家族中尚无AS再发的报告。因此,先证者ID可能代表母亲卵子形成过程中15q11.2-q13印记出现了新发缺陷[Buiting et al 1998]。在这些家庭中,先证者的同胞受累风险低于1%。有单个研究报告了一对AS同胞患者,他们存在1-1.5 Mb倒位,将两个IC元件区分开,但并不存在真正的IC缺失 [Buiting et al 2001]。
IV.UBE3A致病性变异可以是遗传或新发 [Kishino et al 1997, Matsuura et al 1997, Lossie et al 2001, Bürger et al 2002]。大约30%的致病性变异源自遗传。应进行家族性研究,以明确是母系遗传或不是父系遗传[Sadikovic et al 2014]。另外,一些UBE3A致病性变异的体细胞和 胚系嵌合病例已经引起了注意[Malzac et al 1998, Hosoki et al 2005]。如果先证者的母亲存在UBE3A致病性变异,则同胞的受累风险为50%。
V. 在这种分子分型中,患者具有AS的临床特征,但引起AS的遗传机制尚未确定。
先证者的后代。迄今为止,仅报道了一例AS患者生育后代[Lossie & Driscoll 1999]。后代的风险应经过正式遗传咨询后进行确定。
先证者的其他家庭成员。如果在先证者的母亲体内鉴定到UBE3A致病性变异、IC缺失或染色体结构重排,或患者父亲体内出现UPD和罗伯逊易位时,则应为携带的父或母的同胞提供遗传咨询,并选择遗传学检测:
对高危妊娠进行产前诊断和植入前遗传诊断 (PGD)时,需要事先确定家族中的致病机制。
高风险. 15q11.2-q13区域中所有导致AS的已知分子遗传改变(比如分子类别Ia,Ib,IIa,IIb,IIIa,IIIb,IV;参见表 2),经绒毛膜取样(通常在妊娠第10-12周进行)或羊膜穿刺术(通常在妊娠第15-18周进行)获得胎儿细胞,通过DNA和/或染色体/FISH分析进行产前检测 [Kubota et al 1996, Glenn et al 2000, Chang et al 2014]。
理论上可以利用CVS获得的细胞进行DNA甲基化分析(5-7Mb缺失、UPD和IC缺陷)[Kubota et al 1996, Glenn et al 2000]。然而,因为胎盘来源的细胞其DNA甲基化水平相对较低,使用DNA甲基化分析进行产前检测的少数临床实验室更倾向于使用羊水细胞。CVS用于 FISH分析,IC 缺失分析和UBE3A序列分析,在技术上应该是可能的。
由于先证者的分子缺陷类型不同,其同胞的再发风险和所使用的 分子遗传学检测技术存在差异(见建立诊断),产前检查应在先证者遗传机制确定后并且已告知该夫妇未出生孩子的风险之后进行。
- 备注:孕龄以孕周表示,为从正常末次月经第一天算起或通过超声测量计算。
- 低风险。对于无AS家族史的低危妊娠,在以下情况下需要考虑AS:
- 如果CVS或羊膜穿刺的细胞遗传学分析怀疑存在15q11.2-q13缺失,则需要进行FISH分析或染色体芯片分析(CMA)检测。如果证实存在缺失,可以进行亲本来源检测 [Kubota et al 1996, Glenn et al 2000] ,以确定缺失是否来自母源(胎儿为AS)或父源(胎儿为PWS)。
- 如果在CVS上检测到15-三体或嵌合型15-三体,同时如果羊膜穿刺结果显示存在46条染色体,则必须考虑可能由于丢失一条亲本15号染色体的三体性自救所致的AS(父系UPD)或PWS(母系UPD)。在这种情况下,可以进行羊水细胞的亲源(DNA)研究。
其他
辅助生殖技术(ART). 已经证明体外受精(IVF)和胞质内精子注射(ICSI)会增加后代罹患某些 印记疾病的机会,如:Beckwith-Wiedemann综合征。然而,最近的研究表明AS和IVF或ICSI之间并无显著相关性[Vermeiden & Bernardus 2013]。
生育. 荷兰和德国的研究表明,生育问题与AS发生率之间存在相关性。生育AS患儿之前经历过生育问题的夫妇,占比为19%至25%。而在英国咨询的家庭中,生育问题和AS之间并不存在正相关性 [Ludwig et al 2005, Doornbos et al 2007, Vermeiden & Bernardus 2013]。
来源
GeneReviews的工作人员选择如下具体疾病和/或支持型社会组织和/或注册机构,造福这种疾病的个体及其家属。 GeneReviews对其他组织提供的信息并不负责。有关选择标准的信息,请点击此处。
- Angelman Syndrome Foundation, Inc. (ASF)4255 Westbrook DriveSuite 219Aurora IL 60504电话 : 800-432-6435 (toll-free); 630-978-4245传真: 630-978-7408邮箱: info@angelman.org
- Foundation for Angelman Syndrome Therapeutics (FAST)PO Box 608Downers Grove IL 60515电话: 630-852-FAST; 866-783-0078传真: 630-852-3270邮箱: info@CureAngelman.org
- My46 Trait Profile
- National Library of Medicine Genetics Home Reference
- NCBI Genes and Disease
- American Epilepsy Society (AES)
- Epilepsy Foundation8301 Professional Place EastSuite 200Landover MD 20785-7223电话: 800-332-1000 (toll-free)邮件: ContactUs@efa.org
分子遗传
分子遗传学和OMIM表中的信息可能与GeneReview中的其他信息有所不同:表中可能会包含更多的最新信息。 -ED。
表A
天使综合征:基因和数据库
| 基因 | 染色体位点 | 蛋白 | 位点特异数据库 | HGMD ClinVar | |||
|---|---|---|---|---|---|---|---|
| UBE3A | 15q11.2 | 泛素蛋白连接酶E3A | UBE3A at NGRL, Manchester LOVD | UBE3A UBE3A |
分子遗传学病理机制
基因组印记是哺乳动物中存在的一种现象,印记基因依据亲本来源的不同而存在差异表达。AS的主要特征是由于母源UBE3A 等位基因的表达或功能缺陷所致[Jiang et al 1999, Lossie et al 2001, Nicholls & Knepper 2001]。泛素蛋白连接酶E3A参与泛素化途径,对特定蛋白进行降解。
在人类胎儿大脑和成人额叶皮质中,UBE3A主要为母源表达 [Rougeulle et al 1997, Vu & Hoffman 1997, Herzing et al 2001]。在小鼠脑部特定亚区,包括海马、小脑浦肯野细胞、嗅球的二尖瓣细胞和视皮层检测到母源等位基因特异性表达 [Albrecht et al 1997, Jiang et al 1998, Yashiro et al 2009]。UBE3A等位基因不是全身性表达,但其特异性表达在小鼠和人类大脑神经元中是广泛存在的。来自胎鼠大脑的原代细胞培养已经证明UBE3A 印记仅限于神经元,但神经胶质细胞显示的是双等位基因的表达 [Yamasaki et al 2003]。使用RNA-FISH 进行的研究表明,在淋巴细胞和成纤维细胞中母源UBE3A会优先表达,但亲本等位基因之间的差异表达并不像在大脑中那样显著[Herzing et al 2002]。
UBE3A有一个大的5'CpG岛,但与“PWS 关键区域”中的基因相反,母源和父源等位基因之间的DNA 甲基化没有差异[Lossie et al 2001]。
由于UBE3A基因中不存在差异甲基化区域,因此有人提出父源表达的反义转录产物可以间接调节UBE3A基因的印记表达[Rougeulle et al 1998]。Runte等[2001] 研究表明,在AS/PWS 区域存在较长的SNURF-SNRPN正义/UBE3A反义RNA转录单元,从SNURF-SNRPN IC区域开始至少延伸至UBE3A基因的5'末端,跨越长度约460kb。有人认为这种UBE3A反义转录物阻断了父源UBE3A的表达。
基因结构.UBE3A的基因组DNA长约120kb,含有16个外显子。5'非翻译区(UTR)包含数千个碱基,从翻译起始位点上游延伸到6-9个外显子 [Kishino et al 1997, Vu & Hoffman 1997, Yamamoto et al 1997, Kishino & Wagstaff 1998],而3' UTR延伸了2.0 kb [Kishino & Wagstaff 1998]。迄今为止,5'UTR的可变剪接产生了9种成熟转录本和2种不成熟转录本 [Kishino et al 1997, Vu & Hoffman 1997, Yamamoto et al 1997, Kishino & Wagstaff 1998],这些转录本会被翻译成三种不同的蛋白质异构体。不同蛋白质异构体的功能尚不清楚。
- 异构体I(NM_130838.1, NP_570853.1)对应于E6-AP的 开放阅读框架(参见正常基因产物)。
- 异构体II(NM_000462.3, NP_000453.2)含有另外的20个氨基酸。
- 异构体III(NM_130839.2, NP_570854.1)在氨基末端还有另外的23个氨基酸。
致病性变异
- 15q11.2-q13缺失(65%-75%)。大多数引发AS的15q11.1-q13缺失会涉及三个染色体断点(近端BP1,BP2和远端BP3)。大约缺失5-7Mb[Knoll et al 1990, Amos-Landgraf et al 1999, Christian et al 1999](见图 2)。小于10%的AS患者,其缺失可能有从BP1/BP2区域延伸到更远端的BP4或BP5区域位置(见图 2)[Sahoo et al 2007]。
- BP1、BP2和BP3区域的特征为低拷贝重复区域((low-copy repeat region, LCRs),主要包含了来自祖先的HECT 结构域和RCC1结构域蛋白2(HERC2)基因的重复 [Pujana et al 2002]。BP3远端的BP位点包含其他LCRs(例如,无HERC2重复),为15号 染色体衍生的重复DNA元件。
- 备注:位于经典缺失区域两侧的微缺失、包括BP1和BP2之间 [Doornbos et al 2009],BP3和BP4之间[Rosenfeld et al 2011]以及15q13.3更远端区域的 微缺失综合征 [Masurel-Paulet et al 2010] 已有描述。然而,因为具有这些缺失的个体并不具有AS的特征,因此这些缺失不应被认作AS的致病性等位基因。
- 15号染色体的父源单亲二体性(3%-7%)。 与PWS相比,AS的父源UPD更有可能发生在合子(受精卵)形成之后[Robinson et al 2000]。减数分裂期形成的父源UPD也有发生,但不像PWS的母源UPD发生在减数分裂期那么常见。
- 染色体15的父系单亲本二体(3%-7%)。与PWS相比,在AS中观察到的父本UPD在起源上最可能是合子后的[Robinson et al . 2000]。有减数分裂起源的父系UPD确实发生,但这种机制比与PWS相关的母系UPD更不常见。
- 印记缺陷(3%)。 这部分AS患者存在IC缺陷,在配子发生过程中破坏了正常印记的重置。尽管这些个体具有双亲遗传的15号染色体 ,但是母源区域的15q11.2-q13呈现父源表观遗传,使得该区域由母源表达的基因出现转录缺陷[Glenn et al 1993, Buiting et al 2001, Buiting et al 2003]。
- 这些缺失(以及与PWS相关的IC缺失)图谱揭示了两个缺失重叠的小区域(SRO),定义为IC区域中的两个关键元件:AS-SRO和PWS-SRO [Buiting et al 1995] (见图 2)。PWS-SRO的大小为4.3kb,与SNURF-SNRPN外显子1/启动子区重叠 [Ohta et al 1999]。在AS个体中发现的IC缺失会对着丝粒端SNURF-SNRPN启动子/外显子 1区域影响更明显。在IC缺失的AS个体(AS-SRO)的最小重叠区域大小为880 bp,位于SNURF-SNRPN外显子1近端35 kb处[Buiting et al 1999, Horsthemke & Buiting 2008]。 IC缺陷引起的大多数AS患者并不存在AS IC区域的缺失,而是具有干扰IC功能的表观遗传缺陷。
- UBE3A(5%-11%)。已报道了150余种致病性变异,其中60%-70%涉及导致移码突变的小缺失和重复 [Camprubí et al 2009, Stenson et al 2009, Abaied et al 2010, Sadikovic et al 2014]。另外约25%涉及 错义和无义的致病性变异、 剪接突变、大缺失和复杂重排 [Stenson et al 2009, Sadikovic et al 2014]。
- 到目前为止所发现的大多数致病性变异都被预测破坏了HECT连接酶 结构域。外显子9和外显子16编码部分HECT结构域,其致病性变异占很高比例,但由于编码区范围较大,高比例的变异并不代表真正的突变热点。携带温和致病性变异(例如某些错义 突变和非移码缺失或重复)的个体可能会显示部分而非全部的AS相关的临床特征。一些AS患者体内存在UBE3A完全或部分缺失或基因内缺失。某些类型的缺失检测方法可能会检测到一部分缺失(参见表 1) [Lawson-Yuen et al 2006, Sato et al 2007].
有关更多信息,请参阅表 A.
正常基因产物。UBE3A基因产生含有865个氨基酸的E6相关蛋白(E6AP)。E6AP最早被认为是一种与p53结合的蛋白,并可以介导其后者与人乳头瘤病毒E6蛋白结合。这会导致p53肿瘤抑制因子通过泛素蛋白酶体途径进行降解,从而促进宫颈癌的发展 [Huibregtse et al 1991, Huang et al 1999]。E6AP促进活化泛素(一种含76个氨基酸的蛋白质)的转移,使其与靶蛋白共价连接。随后,利用26S蛋白酶体途径识别并降解多聚泛素化的底物。E6AP是E3连接酶HECT(与E6AP 羧基-末端同源)结构域家族成员,共同具有40-kd的保守羧基-末端的催化结构域。E6AP的HECT结构域由外显子9至外显子16编码,是一个双叶结构,在双叶连接处具有一个宽的催化口。E6结合位点由外显子 9编码,接受E2泛素连接酶中泛素的半胱氨酸残基活性位点是由外显子16编码 [Yamamoto et al 1997, Kishino & Wagstaff 1998]。发生于催化口内的致病性变异会干扰泛素-硫酯键的形成。事实上,大多数AS致病性变异发生在HECT结构域区域,预测会影响HECT结构域的功能[Huang et al 1999]。
类固醇受体共激活结构域 位于HECT区域的上游,但其在神经元发育中的作用尚未明确。E6AP似乎具有至少两种独立的功能,因为连接酶区域和HECT结构域对于共激活功能域的功能并不是必需的 [El Hokayem & Nawaz 2014]。
异常基因产物。 UBE3A的破坏可能影响蛋白降解和置换的关键神经元过程,正常情况下该过程可能会由功能性泛素-蛋白酶体系统进行平衡或维持。泛素-蛋白酶体系统对细胞功能至关重要,包括信号转导、细胞周期进程、DNA修复和转录调控 [Ciechanover 1998, Hershko & Ciechanover 1998]。
已经发现了数种E6AP蛋白靶点[Kühne & Banks 1998, Kumar et al 1999, Oda et al 1999, Khan et al 2006, Li et al 2006, Reiter et al 2006, Louria-Hayon et al 2009, Shimoji et al 2009, Margolis et al 2010, Scheiffele & Beg 2010, Gossan et al 2014]。
鸟嘌呤交换蛋白ephexin-5调节EphB受体信号的活性,EphB受体信号对于树突生长非常关键[Margolis et al 2010]。已知Eph受体在突触处富集,对于调节树突棘的密度很重要。通过激活鸟嘌呤核苷酸交换因子(GEFs),EphB受体利用Rho家族的小GTP酶(Rho,Rac和Cdc42)与ephrin配体相互作用,调节树突发育 [Murai & Pasquale 2003]。 目前,尚不清楚当E6AP-靶蛋白相互作用异常时是如何导致AS的。但最近确定的靶点强烈提示E6AP蛋白对于正常突触发育和神经可塑性均至关重要。
参考文献
发表指南/一致性声明
- American Society of Human Genetics/American College of Medical Genetics Test and Technology Transfer Committee. Diagnostic testing for Prader-Willi and Angelman syndromes: Report of the ASHG/ACMG Test and Technology Transfer Committee (pdf). 1996. Available online. Accessed 12-29-16.
- Shaffer LG, Agan N, Goldberg JD, Ledbetter DH, Longshore JW, Cassidy SB. American College of Medical Genetics statement on diagnostic testing for uniparental disomy (pdf). 2001. Available online. Accessed 12-29-16.
- Williams CA, Beaudet AL, Clayton-Smith J, Knoll JH, Kyllerman M, Laan LA, Magenis RE, Moncla A, Schinzel AA, Summers JA, Wagstaff J. Angelman syndrome 2005: updated consensus for diagnostic criteria. 2006. Available online. Accessed 12-29-16. [PubMed: 16470747]
引用文献
- Abaied L, Trabelsi M, Chaabouni M, Kharrat M, Kraoua L, M'rad R, Tebib N, Maazoul F, Chaabouni H. A novel UBE3A truncating mutation in large Tunisian Angelman syndrome pedigree. Am J Med Genet A. 2010;152A:141??6. [PubMed: 20034088]
- Albrecht U, Sutcliffe JS, Cattanach BM, Beechey CV, Armstrong D, Eichele G, Beaudet AL. Imprinted expression of the murine Angelman syndrome gene, Ube3a, in hippocampal and Purkinje neurons. Nat Genet. 1997;17:75??8. [PubMed: 9288101]
- Amos-Landgraf JM, Ji Y, Gottlieb W, Depinet T, Wandstrat AE, Cassidy SB, Driscoll DJ, Rogan PK, Schwartz S, Nicholls RD. Chromosome breakage in the Prader-Willi and Angelman syndromes involves recombination between large, transcribed repeats at proximal and distal breakpoints. Am J Hum Genet. 1999;65:370??86. [PMC free article: PMC1377936] [PubMed: 10417280]
- Arn PH, Williams CA, Zori RT, Driscoll DJ, Rosenblatt DS. Methylenetetrahydrofolate reductase deficiency in a patient with phenotypic findings of Angelman syndrome. Am J Med Genet. 1998;77:198??200. [PubMed: 9605586]
- Bird LM. Angelman syndrome: review of clinical and molecular aspects. Appl Clin Genet. 2014;7:93??104. [PMC free article: PMC4036146] [PubMed: 24876791]
- Boyar FZ, Whitney MM, Lossie AC, Gray BA, Keller KL, Stalker HJ, Zori RT, Geffken G, Mutch J, Edge PJ, Voeller KS, Williams CA, Driscoll DJ. A family with a grand-maternally derived interstitial duplication of proximal 15q. Clin Genet. 2001;60:421??30. [PubMed: 11846734]
- Boyd SG, Cross JH, Dan B (1997) EEG features in Angelman syndrome. Eur J Ped Neurol 1:4, A1.
- Brunetti-Pierri N, Berg JS, Scaglia F, Belmont J, Bacino CA, Sahoo T, Lalani SR, Graham B, Lee B, Shinawi M, Shen J, Kang SH, Pursley A, Lotze T, Kennedy G, Lansky-Shafer S, Weaver C, Roeder ER, Grebe TA, Arnold GL, Hutchison T, Reimschisel T, Amato S, Geragthy MT, Innis JW, Obersztyn E, Nowakowska B, Rosengren SS, Bader PI, Grange DK, Naqvi S, Garnica AD, Bernes SM, Fong CT, Summers A, Walters WD, Lupski JR, Stankiewicz P, Cheung SW, Patel A. Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat Genet. 2008;40:1466??71. [PMC free article: PMC2680128] [PubMed: 19029900]
- Bruni O, Ferri R, D'Agostino G, Miano S, Roccella M, Elia M. Sleep disturbances in Angelman syndrome: a questionnaire study. Brain Dev. 2004;26:233??40. [PubMed: 15130689]
- Buiting K. Prader-Willi syndrome and Angelman syndrome. Am J Med Genet C Semin Med Genet. 2010;154C:365??76. [PubMed: 20803659]
- Buiting K, Barnicoat A, Lich C, Pembrey M, Malcolm S, Horsthemke B. Disruption of the bipartite imprinting center in a family with Angelman syndrome. Am J Hum Genet. 2001;68:1290??4. [PMC free article: PMC1226110] [PubMed: 11283796]
- Buiting K, Dittrich B, Gross S, Lich C, Färber C, Buchholz T, Smith E, Reis A, Bürger J, Nöthen MM, Barth-Witte U, Janssen B, Abeliovich D, Lerer I, van den Ouweland AM, Halley DJ, Schrander-Stumpel C, Smeets H, Meinecke P, Malcolm S, Gardner A, Lalande M, Nicholls RD, Friend K, Schulze A, Matthijs G, Kokkonen H, Hilbert P, Van Maldergem L, Glover G, Carbonell P, Willems P, Gillessen-Kaesbach G, Horsthemke B. Sporadic imprinting defects in Prader-Willi syndrome and Angelman syndrome: implications for imprint-switch models, genetic counseling, and prenatal diagnosis. Am J Hum Genet. 1998;63:170??80. [PMC free article: PMC1377255] [PubMed: 9634532]
- Buiting K, Gross S, Lich C, Gillessen-Kaesbach G, el-Maarri O, Horsthemke B. Epimutations in Prader-Willi and Angelman syndromes: a molecular study of 136 patients with an imprinting defect. Am J Hum Genet. 2003;72:571??7. [PMC free article: PMC1180233] [PubMed: 12545427]
- Buiting K, Lich C, Cottrell S, Barnicoat A, Horsthemke B. A 5-kb imprinting center deletion in a family with Angelman syndrome reduces the shortest region of deletion overlap to 880 bp. Hum Genet. 1999;105:665??6. [PubMed: 10647904]
- Buiting K, Saitoh S, Gross S, Dittrich B, Schwartz S, Nicholls RD, Horsthemke B. Inherited microdeletions in the Angelman and Prader-Willi syndromes define an imprinting centre on human chromosome 15. Nat Genet. 1995;9:395??400. [PubMed: 7795645]
- Bürger J, Horn D, Tonnies H, Neitzel H, Reis A. Familial interstitial 570 kbp deletion of the UBE3A gene region causing Angelman syndrome but not Prader-Willi syndrome. Am J Med Genet. 2002;111:233??7. [PubMed: 12210318]
- Camprubí C, Guitart M, Gabau E, Coll MD, Villatoro S, Oltra S, Roselló M, Ferrer I, Monfort S, Orellana C, Martínez F. Novel UBE3A mutations causing Angelman syndrome: different parental origin for single nucleotide changes and multiple nucleotide deletions or insertions. Am J Med Genet A. 2009;149A:343??8. [PubMed: 19213023]
- Cassidy SB, Dykens E, Williams CA. Prader-Willi and Angelman syndromes: sister imprinted disorders. Am J Med Genet. 2000;97:136??46. [PubMed: 11180221]
- Castro-Gago M, Gómez-Lado C, Eirís-Puñal J, Rodríguez-Mugico VM. Abnormal myelination in Angelman syndrome. Eur J Paediatr Neurol. 2010;14:292. [PubMed: 19720548]
- Chang CW, Hsu HK, Kao CC, Huang JY, Kuo PL. Prenatal diagnosis of Prader-Willi syndrome and Angelman syndrome for fetuses with suspicious deletion of chromosomal region 15q11-q13. Int J Gynaecol Obstet. 2014;125:18??21. [PubMed: 24434231]
- Christian SL, Fantes JA, Mewborn SK, Huang B, Ledbetter DH. Large genomic duplicons map to sites of instability in the Prader-Willi/Angelman syndrome chromosome region (15q11-q13). Hum Mol Genet. 1999;8:1025??37. [PubMed: 10332034]
- Christianson AL, Stevenson RE, van der Meyden CH, Pelser J, Theron FW, van Rensburg PL, Chandler M, Schwartz CE. X linked severe mental retardation, craniofacial dysmorphology, epilepsy, ophthalmoplegia, and cerebellar atrophy in a large South African kindred is localised to Xq24-q27. J Med Genet. 1999;36:759??66. [PMC free article: PMC1734236] [PubMed: 10528855]
- Ciechanover A. The ubiquitin-proteasome pathway: on protein death and cell life. EMBO J. 1998;17:7151??60. [PMC free article: PMC1171061] [PubMed: 9857172]
- Clayton-Smith J. Clinical research on Angelman syndrome in the United Kingdom: observations on 82 affected individuals. Am J Med Genet. 1993;46:12??15. [PubMed: 7684188]
- Clayton-Smith J, Pembrey ME. Angelman syndrome. J Med Genet. 1992;29:412??5. [PMC free article: PMC1015993] [PubMed: 1619637]
- Collinson MN, Roberts SE, Crolla JA, Dennis NR. A familial balanced inverted insertion ins(15)(q15q13q11.2) producing Prader-Willi syndrome, Angelman syndrome and duplication of 15q11.2-q13 in a single family: Importance of differentiation from a paracentric inversion. Am J Med Genet A. 2004;126A:27??32. [PubMed: 15039970]
- Dagli A, Buiting K, Williams CA. Molecular and Clinical Aspects of Angelman Syndrome. Mol Syndromol. 2012;2:100??112. [PMC free article: PMC3366701] [PubMed: 22670133]
- Didden R, Korzilius H, Duker P, Curfs L. Communicative functioning in individuals with Angelman syndrome: a comparative study. Disabil Rehabil. 2004;26:1263??7. [PubMed: 15513724]
- Doornbos M, Sikkema-Raddatz B, Ruijvenkamp CA, Dijkhuizen T, Bijlsma EK, Gijsbers AC, Hilhorst-Hofstee Y, Hordijk R, Verbruggen KT, Kerstjens-Frederikse WS, van Essen T, Kok K, van Silfhout AT, Breuning M, van Ravenswaaij-Arts CM. Nine patients with a microdeletion 15q11.2 between breakpoints 1 and 2 of the Prader-Willi critical region, possibly associated with behavioural disturbances. Eur J Med Genet. 2009;52:108??15. [PubMed: 19328872]
- Doornbos ME, Maas SM, McDonnell J, Vermeiden JP, Hennekam RC. Infertility, assisted reproduction technologies and imprinting disturbances: a Dutch study. Hum Reprod. 2007;22:2476??80. [PubMed: 17586835]
- El Hokayem J, Nawaz Z. E6AP in the brain: one protein, dual function, multiple diseases. Mol Neurobiol. 2014;49:827??39. [PubMed: 24091829]
- Fang P, Lev-Lehman E, Tsai TF, Matsuura T, Benton CS, Sutcliffe JS, Christian SL, Kubota T, Halley DJ, Meijers-Heijboer H, Langlois S, Graham JM Jr, Beuten J, Willems PJ, Ledbetter DH, Beaudet AL. The spectrum of mutations in UBE3A causing Angelman syndrome. Hum Mol Genet. 1999;8:129??35. [PubMed: 9887341]
- Fiumara A, Pittalà A, Cocuzza M, Sorge G. Epilepsy in patients with Angelman syndrome. Ital J Pediatr. 2010;36:31. [PMC free article: PMC2865483] [PubMed: 20398390]
- Fridman C, Varela MC, Kok F, Diament A, Koiffmann CP. Paternal UPD15: further genetic and clinical studies in four Angelman syndrome patients. Am J Med Genet. 2000;92:322??7. [PubMed: 10861661]
- Friez MJ, Jones JR, Clarkson K, Lubs H, Abuelo D, Bier JA, Pai S, Simensen R, Williams C, Giampietro PF, Schwartz CE, Stevenson RE. Recurrent infections, hypotonia, and mental retardation caused by duplication of MECP2 and adjacent region in Xq28. Pediatrics. 2006;118:e1687??95. [PubMed: 17088400]
- Galván-Manso M, Campistol J, Conill J, Sanmarti FX. Analysis of the characteristics of epilepsy in 37 patients with the molecular diagnosis of Angelman syndrome. Epileptic Disord. 2005;7:19??25. [PubMed: 15741136]
- Gentile JK, Tan WH, Horowitz LT, Bacino CA, Skinner SA, Barbieri-Welge R, Bauer-Carlin A, Beaudet AL, Bichell TJ, Lee HS, Sahoo T, Waisbren SE, Bird LM, Peters SU. A neurodevelopmental survey of Angelman syndrome with genotype-phenotype correlations. J Dev Behav Pediatr. 2010;31:592??601. [PMC free article: PMC2997715] [PubMed: 20729760]
- Gilfillan GD, Selmer KK, Roxrud I, Smith R, Kyllerman M, Eiklid K, Kroken M, Mattingsdal M, Egeland T, Stenmark H, Sjøholm H, Server A, Samuelsson L, Christianson A, Tarpey P, Whibley A, Stratton MR, Futreal PA, Teague J, Edkins S, Gecz J, Turner G, Raymond FL, Schwartz C, Stevenson RE, Undlien DE, Strømme P. SLC9A6 mutations cause X-linked mental retardation, microcephaly, epilepsy, and ataxia, a phenotype mimicking Angelman syndrome. Am J Hum Genet. 2008;82:1003??10. [PMC free article: PMC2427207] [PubMed: 18342287]
- Gimelli G, Pujana MA, Patricelli MG, Russo S, Giardino D, Larizza L, Cheung J, Armengol L, Schinzel A, Estivill X, Zuffardi O. Genomic inversions of human chromosome 15q11-q13 in mothers of Angelman syndrome patients with class II (BP2/3) deletions. Hum Mol Genet. 2003;12:849??58. [PubMed: 12668608]
- Giroud M, Daubail B, Khayat N, Chouchane M, Berger E, Muzard E, Medeiros de Bustos E, Thauvin-Robinet C, Faivre L, Masurel A, Darmency-Stamboul V, Huet F, Bejot Y, Giroud M, Moulin T. Angelman syndrome: a case series assessing neurological issues in adulthood. Eur Neurol. 2015;73:119??25. [PubMed: 25472600]
- Gitiaux C, Ceballos-Picot I, Marie S, Valayannopoulos V, Rio M, Verrieres S, Benoist JF, Vincent MF, Desguerre I, Bahi-Buisson N. Misleading behavioural phenotype with adenylosuccinate lyase deficiency. Eur J Hum Genet. 2009;17:133??6. [PMC free article: PMC2985950] [PubMed: 18830228]
- Glenn CC, Deng G, Michaelis RC, Tarleton J, Phelan MC, Surh L, Yang TP, Driscoll DJ. DNA methylation analysis with respect to prenatal diagnosis of the Angelman and Prader-Willi syndromes and imprinting. Prenat Diagn. 2000;20:300??6. [PubMed: 10740202]
- Glenn CC, Nicholls RD, Robinson WP, Saitoh S, Niikawa N, Schinzel A, Horsthemke B, Driscoll DJ. Modification of 15q11-q13 DNA methylation imprints in unique Angelman and Prader-Willi patients. Hum Mol Genet. 1993;2:1377??82. [PubMed: 8242060]
- Gossan NC, Zhang F, Guo B, Jin D, Yoshitane H, Yao A, Glossop N, Zhang YQ, Fukada Y, Meng QJ. The E3 ubiquitin ligase UBE3A is an integral component of the molecular circadian clock through regulating the BMAL1 transcription factor. Nucleic Acids Res. 2014;42:5765??75. [PMC free article: PMC4027211] [PubMed: 24728990]
- Harlalka GV, Baple EL, Cross H, Kuhnle S, Cubillos-Rojas M, Matentzoglu K, Patton MA, Wagner K, Coblentz R, Ford DL, Mackay DJ, Chioza BA, Scheffner M, Rosa JL, Crosby AH. Mutation of HERC2 causes developmental delay with Angelman-like features. J Med Genet. 2013;50:65??73. [PubMed: 23243086]
- Harpey JP, Heron D, Prudent M, Lesourd S, Henry I, Royer-Legrain G, Munnich A, Bonnefont JP. Recurrent meiotic nondisjunction of maternal chromosome 15 in a sibship. Am J Med Genet. 1998;76:103??4. [PubMed: 9508076]
- Harting I, Seitz A, Rating D, Sartor K, Zschocke J, Janssen B, Ebinger F, Wolf NI. Abnormal myelination in Angelman syndrome. Eur J Paediatr Neurol. 2009;13:271??6. [PubMed: 18573670]
- Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425??79. [PubMed: 9759494]
- Herzing LB, Cook EH Jr, Ledbetter DH. Allele-specific expression analysis by RNA-FISH demonstrates preferential maternal expression of UBE3A and imprint maintenance within 15q11- q13 duplications. Hum Mol Genet. 2002;11:1707??18. [PubMed: 12095913]
- Herzing LB, Kim SJ, Cook EH Jr, Ledbetter DH. The human aminophospholipid-transporting ATPase gene ATP10C maps adjacent to UBE3A and exhibits similar imprinted expression. Am J Hum Genet. 2001;68:1501??5. [PMC free article: PMC1226137] [PubMed: 11353404]
- Horsthemke B, Buiting K. Genomic imprinting and imprinting defects in humans. Adv Genet. 2008;61:225??46. [PubMed: 18282508]
- Horsthemke B, Maat-Kievit A, Sleegers E, van den Ouweland A, Buiting K, Lich C, Mollevanger P, Beverstock G, Gillessen-Kaesbach G, Schwanitz G. Familial transloctions involving 15q11-q13 can give rise to interstitial deletions causing Prader-Willi or Angelman syndrome. J Med Genet. 1996;33:848??51. [PMC free article: PMC1050765] [PubMed: 8933339]
- Hosoki K, Takano K, Sudo A, Tanaka S, Saitoh S. Germline mosaicism of a novel UBE3A mutation in Angelman syndrome. Am J Med Genet A. 2005;138A:187??9. [PubMed: 16100729]
- Huang HS, Allen JA, Mabb AM, King IF, Miriyala J, Taylor-Blake B, Sciaky N, Dutton JW Jr, Lee HM, Chen X, Jin J, Bridges AS, Zylka MJ, Roth BL, Philpot BD. Topoisomerase inhibitors unsilence the dormant allele of Ube3a in neurons. Nature. 2011;481:185??9. [PMC free article: PMC3257422] [PubMed: 22190039]
- Huang L, Kinnucan E, Wang G, Beaudenon S, Howley PM, Huibregtse JM, Pavletich NP. Structure of an E6AP-UbcH7 complex: insights into ubiquitination by the E2-E3 enzyme cascade. Science. 1999;286:1321??6. [PubMed: 10558980]
- Huibregtse JM, Scheffner M, Howley PM. A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. EMBO J. 1991;10:4129??35. [PMC free article: PMC453163] [PubMed: 1661671]
- Jiang Y, Lev-Lehman E, Bressler J, Tsai TF, Beaudet AL. Genetics of Angelman syndrome. Am J Hum Genet. 1999;65:1??6. [PMC free article: PMC1378067] [PubMed: 10364509]
- Jiang YH, Armstrong D, Albrecht U, Atkins CM, Noebels JL, Eichele G, Sweatt JD, Beaudet AL. Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron. 1998;21:799??811. [PubMed: 9808466]
- Khan OY, Fu G, Ismail A, Srinivasan S, Cao X, Tu Y, Lu S, Nawaz Z. Multifunction steroid receptor coactivator, E6-associated protein, is involved in development of the prostate gland. Mol Endocrinol. 2006;20:544??59. [PubMed: 16254014]
- Kishino T, Lalande M, Wagstaff J. UBE3A/E6-AP mutations cause Angelman syndrome. Nat Genet. 1997;15:70??3. [PubMed: 8988171]
- Kishino T, Wagstaff J. Genomic organization of the UBE3A/E6-AP gene and related pseudogenes. Genomics. 1998;47:101??7. [PubMed: 9465301]
- Knoll JH, Nicholls RD, Magenis RE, Glatt K, Graham JM Jr, Kaplan L, Lalande M. Angelman syndrome: three molecular classes identified with chromosome 15q11q13-specific DNA markers. Am J Hum Genet. 1990;47:149??54. [PMC free article: PMC1683759] [PubMed: 1971993]
- Kokkonen H, Leisti J. An unexpected recurrence of Angelman syndrome suggestive of maternal germ-line mosaicism of del(15)(q11q13) in a Finnish family. Hum Genet. 2000;107:83??5. [PubMed: 10982040]
- Korff CM, Kelley KR, Nordli DR Jr. Notched delta, phenotype, and Angelman syndrome. J Clin Neurophysiol. 2005;22:238??43. [PubMed: 16093895]
- Kubota T, Aradhya S, Macha M, Smith AC, Surh LC, Satish J, Verp MS, Nee HL, Johnson A, Christan SL, Ledbetter DH. Analysis of parent of origin specific DNA methylation at SNRPN and PW71 in tissues: implication for prenatal diagnosis. J Med Genet. 1996;33:1011??4. [PMC free article: PMC1050812] [PubMed: 9004133]
- Kühne C, Banks L. E3-ubiquitin ligase/E6-AP links multicopy maintenance protein 7 to the ubiquitination pathway by a novel motif, the L2G box. J Biol Chem. 1998;273:34302??9. [PubMed: 9852095]
- Kumar S, Talis AL, Howley PM. Identification of HHR23A as a substrate for E6-associated protein- mediated ubiquitination. J Biol Chem. 1999;274:18785??92. [PubMed: 10373495]
- Kuroda Y, Ohashi I, Saito T, Nagai J, Ida K, Naruto T, Wada T, Kurosawa K. Deletion of UBE3A in brothers with Angelman syndrome at the breakpoint with an inversion at 15q11.2. Am J Med Genet A. 2014;164A:2873??8. [PubMed: 25099823]
- Larson AM, Shinnick JE, Shaaya EA, Thiele EA, Thibert RL. Angelman syndrome in adulthood. Am J Med Genet A. 2015;167A:331??44. [PMC free article: PMC5534346] [PubMed: 25428759]
- Lawson-Yuen A, Wu BL, Lip V, Sahoo T, Kimonis V. Atypical cases of Angelman syndrome. Am J Med Genet A. 2006;140:2361??4. [PubMed: 17036311]
- Li L, Li Z, Howley PM, Sacks DB. E6AP and calmodulin reciprocally regulate estrogen receptor stability. J Biol Chem. 2006;281:1978??85. [PubMed: 16314411]
- Lossie AC, Driscoll DJ. Transmission of Angelman syndrome by an affected mother. Genet Med. 1999;1:262??6. [PubMed: 11258627]
- Lossie AC, Whitney MM, Amidon D, Dong HJ, Chen P, Theriaque D, Hutson A, Nicholls RD, Zori RT, Williams CA, Driscoll DJ. Distinct phenotypes distinguish the molecular classes of Angelman syndrome. J Med Genet. 2001;38:834??45. [PMC free article: PMC1734773] [PubMed: 11748306]
- Louria-Hayon I, Alsheich-Bartok O, Levav-Cohen Y, Silberman I, Berger M, Grossman T, Matentzoglu K, Jiang YH, Muller S, Scheffner M, Haupt S, Haupt Y. E6AP promotes the degradation of the PML tumor suppressor. Cell Death Differ. 2009;16:1156??66. [PubMed: 19325566]
- Low D, Chen KS. UBE3A regulates MC1R expression: a link to hypopigmentation in Angelman syndrome. Pigment Cell Melanoma Res. 2011;24:944??52. [PubMed: 21733131]
- Ludwig M, Katalinic A, Gross S, Sutcliffe A, Varon R, Horsthemke B. Increased prevalence of imprinting defects in patients with Angelman syndrome born to subfertile couples. J Med Genet. 2005;42:289??91. [PMC free article: PMC1736039] [PubMed: 15805153]
- Lugtenberg D, Kleefstra T, Oudakker AR, Nillesen WM, Yntema HG, Tzschach A, Raynaud M, Rating D, Journel H, Chelly J, Goizet C, Lacombe D, Pedespan JM, Echenne B, Tariverdian G, O'Rourke D, King MD, Green A, van Kogelenberg M, Van Esch H, Gecz J, Hamel BC, van Bokhoven H, de Brouwer AP. Structural variation in Xq28: MECP2 duplications in 1% of patients with unexplained XLMR and in 2% of male patients with severe encephalopathy. Eur J Hum Genet. 2009;17:444??53. [PMC free article: PMC2986218] [PubMed: 18985075]
- Malzac P, Webber H, Moncla A, Graham JM, Kukolich M, Williams C, Pagon RA, Ramsdell LA, Kishino T, Wagstaff J. Mutation analysis of UBE3A in Angelman syndrome patients. Am J Hum Genet. 1998;62:1353??60. [PMC free article: PMC1377156] [PubMed: 9585605]
- Margolis SS, Salogiannis J, Lipton DM, Mandel-Brehm C, Wills ZP, Mardinly AR, Hu L, Greer PL, Bikoff JB, Ho HY, Soskis MJ, Sahin M, Greenberg ME. EphB-mediated degradation of the RhoA GEF Ephexin5 relieves a developmental brake on excitatory synapse formation. Cell. 2010;143:442??55. [PMC free article: PMC2967209] [PubMed: 21029865]
- Masurel-Paulet A, Andrieux J, Callier P, Cuisset JM, Le Caignec C, Holder M, Thauvin-Robinet C, Doray B, Flori E, Alex-Cordier MP, Beri M, Boute O, Delobel B, Dieux A, Vallee L, Jaillard S, Odent S, Isidor B, Beneteau C, Vigneron J, Bilan F, Gilbert-Dussardier B, Dubourg C, Labalme A, Bidon C, Gautier A, Pernes P, Pinoit JM, Huet F, Mugneret F, Aral B, Jonveaux P, Sanlaville D, Faivre L. Delineation of 15q13.3 microdeletions. Clin Genet. 2010;78:149??61. [PubMed: 20236110]
- Matsuura T, Sutcliffe JS, Fang P, Galjaard RJ, Jiang YH, Benton CS, Rommens JM, Beaudet AL. De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat Genet. 1997;15:74??7. [PubMed: 8988172]
- Meng L, Ward AJ, Chun S, Bennett CF, Beaudet AL, Rigo F. Towards a therapy for Angelman syndrome by targeting a long non-coding RNA. Nature. 2015;518:409??12. [PMC free article: PMC4351819] [PubMed: 25470045]
- Mertz LG, Christensen R, Vogel I, Hertz JM, Nielsen KB, Gronskov K, Ostergaard JR. Angelman syndrome in Denmark. birth incidence, genetic findings, and age at diagnosis. Am J Med Genet A. 2013;161A:2197??203. [PubMed: 23913711]
- Murai KK, Pasquale EB. 'Eph'ective signaling: forward, reverse and crosstalk. J Cell Sci. 2003;116:2823??32. [PubMed: 12808016]
- Nazlican H, Zeschnigk M, Claussen U, Michel S, Boehringer S, Gillessen-Kaesbach G, Buiting K, Horsthemke B. Somatic mosaicism in patients with Angelman syndrome and an imprinting defect. Hum Mol Genet. 2004;13:2547??55. [PubMed: 15385437]
- Nicholls RD, Knepper JL. Genome organization, function, and imprinting in Prader-Willi and Angelman syndromes. Annu Rev Genomics Hum Genet. 2001;2:153??75. [PubMed: 11701647]
- Nygren AO, Ameziane N, Duarte HM, Vijzelaar RN, Waisfisz Q, Hess CJ, Schouten JP, Errami A. Methylation-specific MLPA (MS-MLPA): simultaneous detection of CpG methylation and copy number changes of up to 40 sequences. Nucleic Acids Res. 2005;33:e128. [PMC free article: PMC1187824] [PubMed: 16106041]
- Oda H, Kumar S, Howley PM. Regulation of the Src family tyrosine kinase Blk through E6AP-mediated ubiquitination. Proc Natl Acad Sci U S A. 1999;96:9557??62. [PMC free article: PMC22247] [PubMed: 10449731]
- Ohta T, Buiting K, Kokkonen H, McCandless S, Heeger S, Leisti H, Driscoll DJ, Cassidy SB, Horsthemke B, Nicholls RD. Molecular mechanism of angelman syndrome in two large families involves an imprinting mutation. Am J Hum Genet. 1999;64:385??96. [PMC free article: PMC1377749] [PubMed: 9973277]
- Peippo MM, Simola KO, Valanne LK, Larsen AT, Kähkönen M, Auranen MP, Ignatius J. Pitt-Hopkins syndrome in two patients and further definition of the phenotype. Clin Dysmorphol. 2006;15:47??54. [PubMed: 16531728]
- Pelc K, Boyd SG, Cheron G, Dan B. Epilepsy in Angelman syndrome. Seizure. 2008a;17:211??7. [PubMed: 17904873]
- Pelc K, Cheron G, Boyd SG, Dan B. Are there distinctive sleep problems in Angelman syndrome? Sleep Med. 2008b;9:434??41. [PubMed: 17765640]
- Peters SU, Bird LM, Kimonis V, Glaze DG, Shinawi LM, Bichell TJ, Barbieri-Welge R, Nespeca M, Anselm I, Waisbren S, Sanborn E, Sun Q, O'Brien WE, Beaudet AL, Bacino CA. Double-blind therapeutic trial in Angelman syndrome using betaine and folic acid. Am J Med Genet A. 2010;152A:1994??2001. [PMC free article: PMC3172130] [PubMed: 20635355]
- Precht KS, Lese CM, Spiro RP, Huttenlocher PR, Johnston KM, Baker JC, Christian SL, Kittikamron K, Ledbetter DH. Two 22q telomere deletions serendipitously detected by FISH. J Med Genet. 1998;35:939??42. [PMC free article: PMC1051488] [PubMed: 9832042]
- Procter M, Chou LS, Tang W, Jama M, Mao R. Molecular diagnosis of Prader-Willi and Angelman syndromes by methylation-specific melting analysis and methylation-specific multiplex ligation-dependent probe amplification. Clin Chem. 2006;52:1276??83. [PubMed: 16690734]
- Pujana MA, Nadal M, Guitart M, Armengol L, Gratacos M, Estivill X. Human chromosome 15q11-q14 regions of rearrangements contain clusters of LCR15 duplicons. Eur J Hum Genet. 2002;10:26??35. [PubMed: 11896453]
- Ramsden SC, Clayton-Smith J, Birch R, Buiting K. Practice guidelines for the molecular analysis of Prader-Willi and Angelman syndromes. BMC Med Genet. 2010;11:70. [PMC free article: PMC2877670] [PubMed: 20459762]
- Reiter LT, Seagroves TN, Bowers M, Bier E. Expression of the Rho-GEF Pbl/ECT2 is regulated by the UBE3A E3 ubiquitin ligase. Hum Mol Genet. 2006;15:2825??35. [PMC free article: PMC3742451] [PubMed: 16905559]
- Robinson WP, Christian SL, Kuchinka BD, Penaherrera MS, Das S, Schuffenhauer S, Malcolm S, Schinzel AA, Hassold TJ, Ledbetter DH. Somatic segregation errors predominantly contribute to the gain or loss of a paternal chromosome leading to uniparental disomy for chromosome 15. Clin Genet. 2000;57:349??58. [PubMed: 10852369]
- Rosenfeld JA, Stephens LE, Coppinger J, Ballif BC, Hoo JJ, French BN, Banks VC, Smith WE, Manchester D, Tsai AC, Merrion K, Mendoza-Londono R, Dupuis L, Schultz R, Torchia B, Sahoo T, Bejjani B, Weaver DD, Shaffer LG. Deletions flanked by breakpoints 3 and 4 on 15q13 may contribute to abnormal phenotypes. Eur J Hum Genet. 2011;19:547??54. [PMC free article: PMC3083619] [PubMed: 21248749]
- Rougeulle C, Cardoso C, Fontes M, Colleaux L, Lalande M. An imprinted antisense RNA overlaps UBE3A and a second maternally expressed transcript. Nat Genet. 1998;19:15??6. [PubMed: 9590281]
- Rougeulle C, Glatt H, Lalande M. The Angelman syndrome candidate gene, UBE3A/E6-AP, is imprinted in brain. Nat Genet. 1997;17:14??5. [letter] [PubMed: 9288088]
- Rubin DI, Patterson MC, Westmoreland BF, Klass DW. Angelman's syndrome: clinical and electroencephalographic findings. Electroencephalogr Clin Neurophysiol. 1997;102:299??302. [PubMed: 9146490]
- Runte M, Huttenhofer A, Gross S, Kiefmann M, Horsthemke B, Buiting K. The IC-SNURF-SNRPN transcript serves as a host for multiple small nucleolar RNA species and as an antisense RNA for UBE3A. Hum Mol Genet. 2001;10:2687??700. [PubMed: 11726556]
- Sadikovic B, Fernandes P, Zhang VW, Ward PA, Miloslavskaya I, Rhead W, Rosenbaum R, Gin R, Roa B, Fang P. Mutation Update for UBE3A variants in Angelman syndrome. Hum Mutat. 2014;35:1407??17. [PubMed: 25212744]
- Sahoo T, Bacino CA, German JR, Shaw CA, Bird LM, Kimonis V, Anselm I, Waisbren S, Beaudet AL, Peters SU. Identification of novel deletions of 15q11q13 in Angelman syndrome by array-CGH: molecular characterization and genotype-phenotype correlations. Eur J Hum Genet. 2007;15:943??9. [PubMed: 17522620]
- Sahoo T, Peters SU, Madduri NS, Glaze DG, German JR, Bird LM, Barbieri-Welge R, Bichell TJ, Beaudet AL, Bacino CA. Microarray based comparative genomic hybridization testing in deletion bearing patients with Angelman syndrome: genotype-phenotype correlations. J Med Genet. 2006;43:512??6. [PMC free article: PMC2564536] [PubMed: 16183798]
- Saitoh S, Wada T, Okajima M, Takano K, Sudo A, Niikawa N. Uniparental disomy and imprinting defects in Japanese patients with Angelman syndrome. Brain Dev. 2005;27:389??91. [PubMed: 16023557]
- Sánchez J, Fernández R, Madruga M, Bernabeu-Wittel J, Antiñolo G, Borrego S. Somatic and germ-line mosaicism of deletion 15q11.2-q13 in a mother of dyzigotic twins with Angelman syndrome. Am J Med Genet A. 2014;164A:370??6. [PubMed: 24311297]
- Sato K, Iwakoshi M, Shimokawa O, Sakai H, Ohta T, Saitoh S, Miyake N, Niikawa N, Harada N, Saitsu H, Mizuguchi T, Matsumoto N. Angelman syndrome caused by an identical familial 1,487-kb deletion. Am J Med Genet A. 2007;143A:98??101. [PubMed: 17152063]
- Scheiffele P, Beg AA. Neuroscience: Angelman syndrome connections. Nature. 2010;468:907??8. [PubMed: 21164477]
- Schroer RJ, Holden KR, Tarpey PS, Matheus MG, Griesemer DA, Friez MJ, Fan JZ, Simensen RJ, Strømme P, Stevenson RE, Stratton MR, Schwartz CE. Natural history of Christianson syndrome. Am J Med Genet A. 2010;152A:2775??83. [PMC free article: PMC3698558] [PubMed: 20949524]
- Sharkey FH, Morrison N, Murray R, Iremonger J, Stephen J, Maher E, Tolmie J, Jackson AP. 17q21.31 microdeletion syndrome: further expanding the clinical phenotype. Cytogenet Genome Res. 2009;127:61??6. [PubMed: 20110647]
- Shimoji T, Murakami K, Sugiyama Y, Matsuda M, Inubushi S, Nasu J, Shirakura M, Suzuki T, Wakita T, Kishino T, Hotta H, Miyamura T, Shoji I. Identification of annexin A1 as a novel substrate for E6AP-mediated ubiquitylation. J Cell Biochem. 2009;106:1123??35. [PubMed: 19204938]
- Spiegel EK, Colman RF, Patterson D. Adenylosuccinate lyase deficiency. Mol Genet Metab. 2006;89:19??31. [PubMed: 16839792]
- Stalker HJ, Williams CA. Genetic counseling in Angelman syndrome: the challenges of multiple causes. Am J Med Genet. 1998;77:54??9. [PubMed: 9557895]
- Steffenburg S, Gillberg CL, Steffenburg U, Kyllerman M. Autism in Angelman syndrome: a population-based study. Pediatr Neurol. 1996;14:131??6. [PubMed: 8703225]
- Stenson PD, Mort M, Ball EV, Howells K, Phillips AD, Thomas NS, Cooper DN. The Human Gene Mutation Database: 2008 update. Genome Med. 2009;1:13. [PMC free article: PMC2651586] [PubMed: 19348700]
- Takano K, Lyons M, Moyes C, Jones J, Schwartz CE. Two percent of patients suspected of having Angelman syndrome have TCF4 mutations. Clin Genet. 2010;78:282??8. [PubMed: 20184619]
- Tan WH, Bacino CA, Skinner SA, Anselm I, Barbieri-Welge R, Bauer-Carlin A, Beaudet AL, Bichell TJ, Gentile JK, Glaze DG, Horowitz LT, Kothare SV, Lee HS, Nespeca MP, Peters SU, Sahoo T, Sarco D, Waisbren SE, Bird LM. Angelman syndrome: Mutations influence features in early childhood. Am J Med Genet A. 2011;155A:81??90. [PMC free article: PMC3563320] [PubMed: 21204213]
- Tan WH, Bird LM, Thibert RL, Williams CA. If not Angelman, what is it? A review of Angelman-like syndromes. Am J Med Genet A. 2014;164A:975??92. [PubMed: 24779060]
- Thibert RL, Conant KD, Braun EK, Bruno P, Said RR, Nespeca MP, Thiele EA. Epilepsy in Angelman syndrome: a questionnaire-based assessment of the natural history and current treatment options. Epilepsia. 2009;50:2369??76. [PubMed: 19453717]
- Thibert RL, Larson AM, Hsieh DT, Raby AR, Thiele EA. Neurologic manifestations of Angelman syndrome. Pediatr Neurol. 2013;48:271??9. [PubMed: 23498559]
- Thibert RL, Pfeifer HH, Larson AM, Raby AR, Reynolds AA, Morgan AK, Thiele EA. Low glycemic index treatment for seizures in Angelman syndrome. Epilepsia. 2012;53:1498??502. [PubMed: 22779920]
- Valente KD, Varela MC, Koiffmann CP, Andrade JQ, Grossmann R, Kok F, Marques-Dias MJ. Angelman syndrome caused by deletion: a genotype-phenotype correlation determined by breakpoint. Epilepsy Res. 2013;105:234??9. [PubMed: 23352739]
- van Bon BW, Koolen DA, Brueton L, McMullan D, Lichtenbelt KD, Adès LC, Peters G, Gibson K, Moloney S, Novara F, Pramparo T, Dalla Bernardina B, Zoccante L, Balottin U, Piazza F, Pecile V, Gasparini P, Guerci V, Kets M, Pfundt R, de Brouwer AP, Veltman JA, de Leeuw N, Wilson M, Antony J, Reitano S, Luciano D, Fichera M, Romano C, Brunner HG, Zuffardi O, de Vries BB. The 2q23.1 microdeletion syndrome: clinical and behavioural phenotype. Eur J Hum Genet. 2010;18:163??70. [PMC free article: PMC2987180] [PubMed: 19809484]
- Van Esch H, Bauters M, Ignatius J, Jansen M, Raynaud M, Hollanders K, Lugtenberg D, Bienvenu T, Jensen LR, Gecz J, Moraine C, Marynen P, Fryns JP, Froyen G. Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am J Hum Genet. 2005;77:442??53. [PMC free article: PMC1226209] [PubMed: 16080119]
- Varela MC, Kok F, Otto PA, Koiffmann CP. Phenotypic variability in Angelman syndrome: comparison among different deletion classes and between deletion and UPD subjects. Eur J Hum Genet. 2004;12:987??92. [PubMed: 15470370]
- Vermeiden JP, Bernardus RE. Are imprinting disorders more prevalent after human in vitro fertilization or intracytoplasmic sperm injection? Fertil Steril. 2013;99:642??51. [PubMed: 23714438]
- Vu TH, Hoffman AR. Imprinting of the Angelman syndrome gene, UBE3A, is restricted to brain. Nat Genet. 1997;17:12??3. [PubMed: 9288087]
- Walz NC. Parent report of stereotyped behaviors, social interaction, and developmental disturbances in individuals with Angelman syndrome. J Autism Dev Disord. 2007;37:940??7. [PubMed: 17019625]
- Watson P, Black G, Ramsden S, Barrow M, Super M, Kerr B, Clayton-Smith J. Angelman syndrome phenotype associated with mutations in MECP2, a gene encoding a methyl CpG binding protein. J Med Genet. 2001;38:224??8. [PMC free article: PMC1734853] [PubMed: 11283202]
- Willemsen MH, Vulto-van Silfhout AT, Nillesen WM, Wissink-Lindhout WM, van Bokhoven H, Philip N, Berry-Kravis EM, Kini U, van Ravenswaaij-Arts CM, Delle Chiaie B, Innes AM, Houge G, Kosonen T, Cremer K, Fannemel M, Stray-Pedersen A, Reardon W, Ignatius J, Lachlan K, Mircher C, Helderman van den Enden PT, Mastebroek M, Cohn-Hokke PE, Yntema HG, Drunat S, Kleefstra T. Update on Kleefstra Syndrome. Mol Syndromol. 2012;2:202??12. [PMC free article: PMC3366700] [PubMed: 22670141]
- Williams CA, Beaudet AL, Clayton-Smith J, Knoll JH, Kyllerman M, Laan LA, Magenis RE, Moncla A, Schinzel AA, Summers JA, Wagstaff J. Angelman syndrome 2005: updated consensus for diagnostic criteria. Am J Med Genet A. 2006;140:413??8. [PubMed: 16470747]
- Williams SR, Mullegama SV, Rosenfeld JA, Dagli AI, Hatchwell E, Allen WP, Williams CA, Elsea SH. Haploinsufficiency of MBD5 associated with a syndrome involving microcephaly, intellectual disabilities, severe speech impairment, and seizures. Eur J Hum Genet. 2010;18:436??41. [PMC free article: PMC2987257] [PubMed: 19904302]
- Yamamoto Y, Huibregtse JM, Howley PM. The human E6-AP gene (UBE3A) encodes three potential protein isoforms generated by differential splicing. Genomics. 1997;41:263??6. [PubMed: 9143503]
- Yamasaki K, Joh K, Ohta T, Masuzaki H, Ishimaru T, Mukai T, Niikawa N, Ogawa M, Wagstaff J, Kishino T. Neurons but not glial cells show reciprocal imprinting of sense and antisense transcripts of Ube3a. Hum Mol Genet. 2003;12:837??47. [PubMed: 12668607]
- Yashiro K, Riday TT, Condon KH, Roberts AC, Bernardo DR, Prakash R, Weinberg RJ, Ehlers MD, Philpot BD. Ube3a is required for experience-dependent maturation of the neocortex. Nat Neurosci. 2009;12:777??83. [PMC free article: PMC2741303] [PubMed: 19430469]
- Zweier C, Peippo MM, Hoyer J, Sousa S, Bottani A, Clayton-Smith J, Reardon W, Saraiva J, Cabral A, Gohring I, Devriendt K, de Ravel T, Bijlsma EK, Hennekam RC, Orrico A, Cohen M, Dreweke A, Reis A, Nurnberg P, Rauch A. Haploinsufficiency of TCF4 causes syndromal mental retardation with intermittent hyperventilation (Pitt-Hopkins syndrome). Am J Hum Genet. 2007;80:994??1001. [PMC free article: PMC1852727] [PubMed: 17436255]
- Zweier C, Thiel CT, Dufke A, Crow YJ, Meinecke P, Suri M, Ala-Mello S, Beemer F, Bernasconi S, Bianchi P, Bier A, Devriendt K, Dimitrov B, Firth H, Gallagher RC, Garavelli L, Gillessen-Kaesbach G, Hudgins L, Kaariainen H, Karstens S, Krantz I, Mannhardt A, Medne L, Mucke J, Kibaek M, Krogh LN, Peippo M, Rittinger O, Schulz S, Schelley SL, Temple IK, Dennis NR, Van der Knaap MS, Wheeler P, Yerushalmi B, Zenker M, Seidel H, Lachmeijer A, Prescott T, Kraus C, Lowry RB, Rauch A. Clinical and mutational spectrum of Mowat-Wilson syndrome. Eur J Med Genet. 2005;48:97??111. [PubMed: 16053902]
Chapter Notes
Author History
Adati I Dagli, MD (2008-present)
Hui-Ja Dong; University of Florida College of Medicine (2003-2005)
Daniel J Driscoll, PhD, MD; University of Florida College of Medicine (1998-2011)
Amy C Lossie, PhD; University of Florida College of Medicine (1998-2003)
Jennifer Mueller, GC, (2015-present)
Charles A Williams, MD (1998-present)
Revision History
- 14 May 2015 (me) Comprehensive update posted live
- 16 June 2011 (me) Comprehensive update posted live
- 5 September 2008 (me) Comprehensive update posted live
- 30 July 2007 (cd) Revision: 靶性性突变分析 no longer available clinically
- 21 February 2007 (cd) Revision: clarification of Genetic Counseling section
- 8 November 2005 (me) Comprehensive update posted to live Web site
- 3 September 2004 (cw) Author revisions
- 29 July 2003 (me) Comprehensive update posted to live Web site
- 2 April 2002 (cw) Author revision
- 21 November 2000 (me) Comprehensive update posted to live Web site
- 15 September 1998 (pb) Review posted to live Web site
- April 1998 (cw) Original submission