
概述
临床特征.
WFS1 Wolfram 综合征谱系障碍 (WFS1-WSSD) 是一种进行性神经退行性疾病,其特征是在16岁前患糖尿病 (DM) 和视神经萎缩 (OA),通常伴有其他内分泌异常、感音神经性听力损失和进行性神经异常(小脑共济失调,周围神经病变、痴呆、精神疾病和泌尿道张力低下)。尽管罹患糖尿病的类型主要是胰岛素依赖型糖尿病,但总的病程较单纯性糖尿病温和。 视神经萎缩通常在十岁以前就开始引起视力显著下降。感音神经性听力损失是先天性耳聋或更轻的,有时表现为进行性的听力损失。
健康管理.
对症治疗: 建议(基于Wolfram综合征的详细临床指南)由多学科医生对以下疾病进行常规治疗:胰岛素依赖型糖尿病;视神经萎缩;听力障碍;日常活动和运动;构音障碍;吞咽困难;内分泌失调;发育迟缓/智力障碍;神经性膀胱功能障碍;精神/行为问题。
监管: 常规随访。以评估持续医疗与护理的有效性以及确定新的疾病表型。
遗传咨询.
WFS1-WSSD的遗传模式是常染色体隐性遗传。如果父母双方均携带杂合的WFS1致病性变异,那么受累个体的同胞都有25%的概率受累,有50%概率为无症状的携带者,25% 的概率不受累且不携带变异。当家族中的 WFS1 致病性变异明确时,可进行有风险亲属的携带者检测、高风险孕妇的产前诊断,也可行胚胎植入前遗传学诊断。
诊断
提示性特征
有以下特征表现和家族史的患者应怀疑为WFS1 Wolfram综合征谱系障碍 (WFS1-WSSD) :
临床表现 [Barrett et al 1995, Urano 2016]:
- 糖尿病 (发病年龄 <15 岁)
- 视神经萎缩 (发病年龄 <15 岁)
- 高频感音神经性听力损失 (有时是先天且严重的)
- 小脑共济失调
- 痴呆/智力残疾 (两者都可能发生,但智力残疾很少见)
- 精神疾病
- 神经源性膀胱功能障碍或膀胱协同障碍
- 其他内分泌的相关表现:
- 中枢性尿崩症
- 青春期发育延迟/缺失;男性性腺功能减退症
- 非自身免疫性甲状腺功能减退症
- 发育迟缓
- 心肌病和结构性先天心脏缺陷
家族史符合常染色体隐性遗传
建立诊断
WFS1-WSSD先证者的确诊依据是分子遗传学检测确认的WFS1双等位基因致病性变异 (见表1)。
根据患者的表型,可选用目的基因检测 (单基因检测或多基因靶向组合检测) 和综合的基因组检测 (外显子组测序, 基因芯片, 基因组测序)。
目的基因检测要求临床医生确定纳入的基因, 而综合的基因组检测则不需要。因为WFS1-WSSD有多种表型,具有提示性特征表现的患者可以使用目的基因检测 (见选项1),而未考虑WFS1-WSSD诊断的患者更有可能通过综合的基因组检测确诊 (见选项2)。
选项1
单基因检测. 首先对WFS1 进行序列分析,以检测基因内小片段的缺失/插入和错义, 无义和剪接位点变异。注:根据所使用的测序方法,可能无法检测到单外显子,多外显子或全基因缺失/重复。如果此方法只检测到一个变异或没有检测到变异,下一步通常是进行基因靶向缺失/重复分析,以检测外显子和全基因缺失/重复;然而,到目前为止,这些变异还没有被确定为WFS1-WSSD的致病性变异。
耳聋多基因组合检测包括了 WFS1和其他相关基因 (见鉴别诊断) 是成本最合理又最可能鉴定出遗传病因的策略,同时减少了意义不确定变异的检出和无法解释对应表型的基因中致病性变异的检出。注意:(1) 靶向组合中包含的基因和每个基因的诊断敏感性在不同的实验室可能存在差异,而且会随着时间而改变。 (2) 有些靶向基因组合中可能不仅仅包含本篇GeneReview中疾病相关的基因。 (3) 在有些实验室,这种靶向组合包含了实验室定制的靶向基因和/或临床医生特定的以表型为依据定制的外显子组分析。(4) 靶向组合应用的方法可能包含了序列分析、缺失/重复分析和/或其他非测序类的检测。
选项2
综合的基因组检测不需要临床医生决定哪些基因需要纳入。 外显子测序是最常用的方法; 基因组测序也是可行的。
如果外显子组测序未能诊断,外显子组芯片 (临床可用的情况下) 可以考虑用于检测序列分析无法检测到的 (多个) 外显子缺失或重复;然而,到目前为止,这些变异还没有被确定为WFS1-WSSD的致病性变异。
关于综合的基因组检测的介绍请点击这里。更多临床医生定制基因组检测的详细信息请点击这里。
Table 1.
表1.
Wolfram综合征谱系障碍 (WFS1) 的分子遗传学检测
| 基因1 | 方法 | 该检测技术检出致病性变异2的占比 |
|---|---|---|
| WFS1 | 序列分析3 | >95%4 |
| 靶向基因的缺失/重复分析5 | 已报道3例6 |
- 1.
- 2.
- 3.
- 4.
Hardy et al [1999], Khanim et al [2001], Smith et al [2004], Chaussenot et al [2015] 和数据来源于人类基因突变数据库 [Stenson et al 2017]。
- 5.
- 6.
三个基因内缺失一个或多个外显子 [Smith et al 2004, Elli et al 2012, Chaussenot et al 2015]。
临床特征
临床描述
典型的常染色体隐性遗传WFS1 Wolfram综合征谱系障碍 (WFS1-WSSD) 特征是在儿童期出现糖尿病、视神经萎缩、听力障碍/耳聋、尿崩症、神经异常和精神异常 (见表2)。
注: 本篇GeneReview主要关注由WFS1双等位基因致病性变异引起的Wolfram综合征谱系病。类Wolfram综合征由WFS1杂合的致病变异引起,并且与常染色体隐性遗传的WFS1-WSSD的临床特征重叠,在相关遗传疾病中进行讨论。
Table 2.
表2.
WFS1-WSSD的选择性特征
| 特征 | 常见 | 不常见 | ||
|---|---|---|---|---|
| 糖尿病 | ● | |||
| 视神经萎缩 | ● | |||
| 感音神经性听力损失 | ● | |||
| 小脑共济失调 | ● | |||
| 自主神经功能障碍 | ● | |||
| 延髓功能障碍 | ● | |||
| 呼吸系统 | ● | |||
| 发育迟缓 (幼儿) | ● | |||
| 智力残疾 (年龄较大的儿童和成人) | ● | |||
| 精神疾病 | ● | |||
| 泌尿道问题 | 功能性:神经性膀胱功能障碍 | ● | ||
| 结构性:上尿路扩张 | ||||
| 肠功能障碍 | ● | |||
| 癫痫 | ● | |||
| 其他内分泌 | 中枢性尿崩症 | ● | ||
| 性腺机能减退 | ● | |||
| 甲状腺功能减退 | ● | |||
| 生长迟缓 | ● | |||
对WFS1及其在不同临床表现中的作用进行了全面的综述 [Tranebjærg 2008]。
WFS1-WSSD是一种进行性神经退行性疾病,以15岁前患糖尿病和视神经萎缩为特征,通常与感音神经性听力损失、进行性神经异常和其他内分泌异常有关。几乎每个器官系统都可能受到影响;然而,由于只有少数已发表的病例进行了广泛而全面的临床检查,WFS1-WSSD对多器官影响的病程发展在很大程度上是未知的。
[Barrett et al 1995]描述了英国29个家系的45名个体的Wolfram综合征的疾病发展情况。20岁时有64%的人存在听力障碍。60%的研究对象(平均年龄16岁,范围为5-32岁)有以下一种或多种疾病:小脑共济失调、周围神经病变、智力残疾、痴呆、精神疾病和尿道无力。寿命大大缩短。在英国、巴基斯坦和阿拉伯/非洲混血血统的家系中,19名先证者中的17名发现了WFS1致病性变异 [Hardy et al 1999]。
糖尿病(DM). 糖尿病的中位发病年龄在10岁以前 (年龄范围 <1-17岁)。几乎所有患者患的糖尿病都是胰岛素依赖型。糖尿病可出现酮症酸中毒;然而,总的来说,Wolfram综合征中的糖尿病病程比单纯性糖尿病温和,微血管疾病 [Cano et al 2007a] 和视网膜微血管病变的发生率较低。
视神经萎缩 (OA). 所有已知的WSSD患者都会出现视神经萎缩。视神经萎缩的病程是进行性的,中位发病年龄在10岁以前;发病8年后,大多数人的视力下降到6/60左右 [Barrett et al 1995]。注:视力6/60是英国“注册盲人”和美国“法定盲人”的定义,这意味着受试者在6米处看到的是正常视力的人在60米处看到的。
WSSD报道的其他眼科发现,但未被证实为该疾病的表型:
- 1例色素性视网膜病变而非视神经萎缩 [Dhalla et al 2006]
- 眼球震颤
约66%的WSSD患者存在感音神经性听力损失,从先天性到更晚发生的耳聋,有时是进行性的感音神经性听力损失。中位发病年龄为12.5岁[Barrett et al 1995]。听力图显示为下降型曲线 [Pennings et al 2004]。在WFS1功能失活变异的个体中,5名女性的听力受损程度明显高于4名男性,据此提出了“激素可能调节听力损失”的猜测 [Pennings et al 2004]。一项多中心研究证实了高频听力下降为主,并且听力损失的进展比较缓慢,但没有证实听力损失程度在性别上的差异 [Plantinga et al 2008]。
随着年龄的增长,言语识别评分的下降比单纯年龄相关性听力下降更明显,这表明随着时间的推移,进行性中枢神经系统的参与也可能是言语困难的原因 [Pennings et al 2004]。
注:虽然经验有限,但前庭功能异常似乎不是WFS1-WSSD的突出特征。在接受评估的6名WFS1-WSSD患者中,只有1人具有前庭反射障碍 [Pennings et al 2004]。平衡问题可能是运动神经异常的结果。
Barrett et al [1995] 研究发现,62%的患者(平均年龄为30岁,范围为5-44岁)在分子诊断确诊之前存在神经病变。然而,关于神经病变类型的发生率非常有限。
目前的经验表明在30多岁表现出神经病变,通常在10多岁发病。
神经病变的临床表现为进行性,是脑干和颅神经受累的广泛脑萎缩的结果 [Barrett et al 1995, Pakdemirli et al 2005, Domenech et al 2006]。45名患者中有8人的脑部MRI异常,表现为广泛性脑萎缩,以小脑、延髓和桥脑部位最为突出;以及视神经和下丘脑后部的信号强度降低 [Barrett et al 1995]。MRI显示的脑萎缩与临床表现之间的相关性并不总是很强 [Ito et al 2007]。
- 在45例研究对象中,有15例发现躯干或步态共济失调 [Barrett et al 1995]。
- 在研究的45名患者中,有5人出现了呼吸暂停,这是一种严重的表现 [Barrett et al 1995]。
- 痴呆症被认为是老年患者更广泛的神经退行性变的一部分。智力残疾并不常见。
- 据观察,自杀行为和需要住院治疗的精神疾病的风险显著增加 [Swift et al 1998]。
其他内分泌检查结果
- 中枢性尿崩症发生率为72%,中位发病年龄为15.5岁。发病年龄的范围很广,可能是由于确诊的延误。
- 性腺机能减退症在男性比女性中发生地更为普遍。它可以表现为促性腺功能低下(即中枢性)或促性腺功能亢进(即继发性性腺功能衰竭)。两种类型的潜在病理机制仍不清楚。女性通常保持着怀孕的能力;文献中描述了大约六次成功妊娠。一位女性没有子宫 [Tranebjærg, 个人观察]。
- 甲状腺功能减退. 发生率尚不清楚。
- 发育迟缓. 大多数成年人身高正常,但发育迟缓并不少见。青春期开始的年龄各不相同。
尿路. 肾流出道扩张(输尿管积水),尿失禁和反复感染是神经源性膀胱疾病的常见症状。在29例患者中,55%患者有此类症状,中位发病年龄为22岁(年龄范围:10-44岁) [Barrett et al 1995]。尿动力学检查显示膀胱不完全排空或完全无张力。
胃肠运动障碍和乳糜泻. 据报道,25%WFS1-WSSD患者出现便秘,慢性腹泻和其他肠道功能障碍,有时是麸质不耐受的结果,该发生率是患有多年糖尿病患者的20倍 [Barera et al 2002, Skovbjerg et al 2005, Liu et al 2006] (见腹腔疾病)。
心肌病. 没有发生率方面的数据。
死亡原因. Barrett et al [1995] 研究报告中的45名患者有10人已经死亡。死亡年龄中位数为30岁。报告显示35岁时的死亡率为65%。然而,值得一提的是,在文献中倾向于报道WSSD最严重的病例,这可能引起偏倚。死亡原因为低血糖昏迷、癫痫持续状态、复发性尿路感染导致的终末期肾病和自杀。3人死于中枢性呼吸衰竭伴脑干萎缩。
神经病理学. 目前,只发表了临床诊断的病例;分子诊断确认病例的神经病理学还未见报道。其中2例表现为小脑、嗅球、视神经、桥脑核、下橄榄核和齿状核的萎缩;耳蜗神经节细胞丢失;脊髓神经元轻度缺失 [Genís et al 1997, Shannon et al 1999]。
基因型-表型关联
WFS1-WSSD 的临床病程是高度可变的,即使在一个家系中也是如此,并且不能从致病性变异的类型或位置来预测。
Cano et al [2007a] 发现WFS1 两个等位基因都是功能失活的致病性变异,易导致糖尿病和视神经萎缩的发病年龄提前。此外,WSSD的表现度更完整,在非错义变异的个体中发生更早。
命名规则
Wolfram综合征有时被命名为DIDMOAD(尿崩症、糖尿病、视神经萎缩和耳聋)。
患病率
超过来自全世界的60个家庭中的90多个人被报道患病 [Khanim et al 2001, Tessa et al 2001, Domènech et al 2002, Colosimo et al 2003, Cryns et al 2003, Simsek et al 2003, van den Ouweland et al 2003, Smith et al 2004, Giuliano et al 2005, Hansen et al 2005, Cano et al 2007b]。
来自英国的一项研究估计,英国儿童WSSD的患病率为1:550,000 [Barrett et al 1995]。
相关遗传(等位基因)疾病
类Wolfram综合征. 在两个具有相同杂合的WFS1错义突变的家系中描述了类Wolfram综合征特征 (见表3) [Eiberg et al 2006, Valéro et al 2008]。在这些家系中,类Wolfram综合征的遗传模式是常染色体显性遗传 (也就是说,只有一个WFS1致病性变异在家系中共分离)。WFS1-WSSD的鉴别诊断应考虑类Wolfram综合征。
注:常染色体显性遗传的类Wolfram综合征的个体/家系会发生(较轻的)视神经萎缩(OA),[Eiberg et al 2006] 的研究表明,在没有OA的情况下,糖尿病(DM)和先天的中度听力损失可能是WFS1杂合致病性变异中未被充分认识的一种表现,以常染色体显性遗传方式出现。
表3.
类Wolfram综合征临床表型
| 类Wolfram综合征表型 | Eiberg et al [2006] 描述的家庭 (岁) | Valéro et al [2008] 描述的家庭 (岁) | ||||
|---|---|---|---|---|---|---|
| F (89) | M(70) | M (67) | F(32) | M (60) | F (81) | |
| DM | – | 葡萄糖耐受降低 | 糖尿病 | – | 非胰岛素依赖型糖尿病 | 非胰岛素依赖型糖尿病 |
| OA | 重度 | 中度 | 中度 | 轻度 | – | 双侧视神经萎缩 |
| 耳聋 | 重度 | 中度 | 中度 | 中度 | 中度 | 轻度 |
DM = 糖尿病; F = 女性; M = 男性;OA = 视神经萎缩
最近报道了一例WFS1 相关的感音神经性听力损失和视神经萎缩 (无糖尿病或尿崩症) 。患者为一名18岁女性,具有WFS1双等位基因的致病性变异。她的父亲有轻微的表型,携带与常染色体显性遗传听力损失相关的WFS1杂合致病性变异 [Lusk et al 2020]。此家庭完善了WFS1致病性变异相关特征谱。
其他未在WFS1-WSSD鉴别诊断中的WFS1等位基因疾病总结见表4。
表4.
WFS1等位基因疾病
| 表型 | MOI | 临床特征/评价 |
|---|---|---|
| 非综合征型 OA1 | AR | |
| 非综合征型Ⅱ型糖尿病2 | AD | |
| 单纯性先天性核白内障3 | AD | 报道称,WFS1的一个错义突变与一个家系的单纯性先天性核白内障有关,其遗传模式是AD。 |
| 非综合征型低频感音神经性听力损失 (DFNA6/14/38) | AD |
|
- 1.
- 2.
- 3.
- 4.
目前尚不清楚在WFS1-WSSD中是高频听力损失而在DFNA6/14/38中是低频听力损失的原因。
鉴别诊断
Wolfram综合征2型(WS2) (OMIM 604928) 是由CISD2双等位基因致病性突变引起的常染色体隐性遗传病。与WFS1-WSSD一样,WS2表现为一系列的临床特征;然而,WS2完整的临床表型谱还没有完全确定,因为已报道患者太少。到目前为止,在WS2患者中已报道了以下临床特征:
- El-Shanti et al [2000], al-Sheyyab et al [2001] 和 Amr et al [2007] 报道4个约旦家系中出现了青少年糖尿病、视神经萎缩、高频感音神经性听力损失、尿路扩张、肾功能受损、性腺功能减退和严重胃肠道溃疡和出血;在一个家系中发现了异常的面部特征 [Amr et al 2007]。
- 意大利和摩洛哥的患者出现了尿崩症、精神异常和不同程度的视神经萎缩 [Mozzillo et al 2014, Rondinelli et al 2015, Rouzier et al 2017]。在这些患者的一部分人中出现了消化性溃疡,皮肤黏膜出血和血小板聚集。
注: 在一个符合Wolfram综合征谱系障碍所有诊断标准但没有WFS1致病性突变的患者中,发现了一个新的CISD2致病性变异 (c.215A>G; p.Asn72Ser) [Rouzier et al 2017]。
听力障碍. 参见遗传性听力损失和耳聋概述。
神经退行性疾病合并糖尿病 (DM). 见表5。
表5.
WFS1-WSSD中神经退行性疾病合并糖尿病的鉴别诊断
| 基因/遗传机制 | 疾病 | MOI | 部分临床特征 | |||
|---|---|---|---|---|---|---|
| 内分泌异常 | 眼部病变 | 听力损失 | 神经系统异常 | |||
| ALMS1 | Alström综合征 | AR | 胰岛素抵抗/2型糖尿病多见于青少年/20岁/其他内分泌异常包括男性的低促性腺激素性性腺功能功能减退,女性的多囊卵巢和甲状腺功能减退。 | 锥杆细胞营养不良表现从出生后到15岁的进行性视力障碍、畏光和眼球震颤;许多人在20岁时没有光感。 | 约70%的个体在10岁以前就患有进行性SNHL。在接近20岁时,听力损失可能变为中度至重度(40-70dB)。 | 20岁左右的女性逼尿肌-尿道协同失调 |
| BBS1 BBS2 BBS4 BBS7 BBS9 BBS10 BBS12 MKKS MKS1 TTC81 | Bardet-Biedl综合征 | AR | 非胰岛素依赖型糖尿病/2型通常出现在青春期或成年期;男性的低促性腺激素性性腺功能功能减退 | 锥杆细胞营养不良;夜盲症通常在7-8岁时出现;发展为法定盲人的平均年龄为15.5岁。 | 约50%的成人SNHL临床症状不明显,只能通过听力检测确认。 | 大多数个体存在显著的学习困难;智商测试显示少数人严重智力障碍。 |
| DMPK | Ⅰ型强直性肌营养不良 (DM1) | AD | 轻度DM1和典型DM1中常伴有糖尿病的发生。 | 轻度DM1和典型DM1伴有白内障的发生。 | NA | 轻度DM1患者出现轻度肌强直(肌肉持续收缩)的情况;典型DM1出现肌无力/消瘦和肌强直的表现。 |
| FXN | Friedreich共济失调 | AR | 30%的患者患有糖尿病 | 视神经萎缩,通常无症状,发生率约为25%。对比度的递进性下降是疾病进展的典型表现。 | 13%的患者患有SNHL | 缓慢进行性共济失调的平均发病年龄为10-15岁(通常小于25岁);构音障碍,肌无力,下肢痉挛,脊柱侧弯,膀胱功能障碍,下肢反射缺失,位置感和振动感丧失。 |
| mtDNA缺失 | Kearns-Sayre综合征(见线粒体DNA缺失综合征) | Mat | 糖尿病,甲状旁腺功能减退和生长激素缺乏 | 色素性视网膜病变及进行性眼外肌麻痹/发病年龄<20岁 | 一些患者出现SNHL | 小脑共济失调;智力受损(ID和/或认知障碍) |
| SLC19A2 | 硫胺素敏感性巨幼细胞性贫血综合征 | AR | 非I型糖尿病;发病年龄从婴儿期到青春期。 | OA (在病例报告中的表型描述中)是常见的。 | 进行性SNHL/一般早发;可以在10至18个月的幼儿身上检测到。SNHL是不可逆的,不能通过维生素 B1治疗加以改善。 | 27%的人在儿童早期被报道有明显的神经功能缺陷,包括中风和局灶性或全身性癫痫。 |
- 1.
列出的基因代表最常见的相关基因;至少有19个基因与Bardet-Biedl综合征有关 (见Bardet-Biedl综合征)。
伴有听觉障碍的视神经萎缩. 见表6。
表6.
WFS1-WSSD的视神经萎缩伴听力障碍的鉴别诊断
| 基因 | 疾病 | MOI | 部分临床特征 | ||
|---|---|---|---|---|---|
| 眼部病变 | 耳聋 | 神经系统异常 | |||
| OPA1 | Ⅰ型视神经萎缩 | AD | 双侧对称视神经苍白,视敏度一般在4-6岁悄然下降;视野缺失;色觉缺陷。视力障碍通常为中度(6/10-2/10),从轻度甚至微不足道到严重(法定盲人的视力<1/20) | 听神经病变到SNHL 的程度从先天、重度和到临床症状不明显1 | 约20%有相关的其他临床特征,特别是神经系统症状。 |
| PRPS1 | 5型腓骨肌萎缩神经病变症(Charcot-Marie-Tooth neuropathy X type 5) | XL | 男性视神经病变;7-20岁开始出现视力障碍。 | 男性早发(语前)双侧极重度SNHL | 男性周围神经病变发病年龄为5-12岁 |
| TIMM8A 2 | 耳聋-肌张力障碍-视神经病变综合征 | XL | 男性的视力在20岁左右从视神经萎缩缓慢进展为视敏度降低。 | 男性儿童早期语前或语后SNHL;女性可能有轻微的听力障碍。 | 在青少年期出现缓慢进行性肌张力障碍或共济失调;40岁左右开始出现痴呆;精神症状(如人格改变,偏执)可能出现在儿童时期和成长过程中。女性可能出现局灶性肌张力障碍。 |
管理
初步诊断后的检查
为明确WFS1 Wolfram 综合征谱系病(WFS1-WSSD)患者的患病程度和需求,推荐的检查总结见表7 (如果诊断时未进行检查) 。
参考 Wolfram综合征临床管理指南,第5页推荐的基线调查.
表7.
初步诊断为WFS1-WSSD后推荐的检查项目
| 相关系统 | 检查 | 备注 | |
|---|---|---|---|
| 糖尿病 | 空腹血糖和糖化血红蛋白A1c | 很少出现糖尿病酮症酸中毒;常见缓解期延长 | |
| 视神经萎缩 | 眼科评估 | 评估:
| |
| 感音神经性听力障碍 | 听力检查 | 包括:
| |
| 运动障碍 | 神经检查包括脑部MRI(如果之前没有做过)和认知评估 | 使用标准化量表建立共济失调基线(SARA,ICARS或BARS)。1评估:
| |
| 转诊到神经肌肉诊室(OT/PT/康复专科医生) | 评估:
| ||
| 自主神经功能障碍 | 获得体位性低血压,无汗症,手足多汗症,便秘,胃轻瘫,低体温和高热。 | ||
| 延髓功能紊乱 | 由言语病理学家评估 | 评估言语障碍(构音障碍)和吞咽障碍(吞咽困难)。 | |
| 呼吸功能 | 多频道睡眠记录 | 中枢性呼吸暂停可继发脑干功能障碍。 | |
| 发育(儿童) | 发育评估 | 包括运动、适应、认知、言语/语言评估,评估用于决定是否开展早期干预/特殊教育。 | |
| 认知障碍(年龄较大的儿童和成人) | 包括:运动和言语/语言评估;一般认知能力 | ||
| 精神病学/行为学 | 神经精神病学的评价 | 年龄>12岁的个体:筛查包括睡眠障碍、多动症、焦虑和/或ASD提示性特征的情况。 | |
| 神经源性膀胱功能障碍 | 膀胱痉挛病史:尿急、尿频、排尿困难、尿失禁、反复感染 | 转诊泌尿科医生;考虑评估泌尿系统动力学,输尿管扩张时的尿路和肾脏成像;肾功能评估。 | |
| 肠功能障碍 | 有便秘史和胃轻瘫 | ||
| 其他内分泌 | 尿崩症 | 评估浓缩尿液的功能 | 晨尿尿量,空腹血浆渗透压,夜间钠离子浓度和晨间血糖稳定。 |
| 性腺机能减退症 | 无迟发性青春期及/或不育症病史 | 转诊内分泌科医生评估原发性性腺功能衰竭和/或低促性腺素性功能减退。 | |
| 甲状腺功能减退症 | 甲状腺功能检测 | 评估甲状腺功能 | |
| 生长迟缓 | 在标准生长图上标出身高、体重和头围 | 确定生长障碍和/或提供参考基线 | |
| 遗传咨询 | 通过遗传学专家2 | 告知患者和家属WFS1-WSSD的性质、MOI和可能的影响,以帮助做出医疗和个人发展相关的决策 | |
| 家庭支持/资源 |
| ||
ABRs = 听觉脑干反应; ADHD = 注意力缺陷/多动障碍; ASD = 自闭症谱系障碍; BARS =简易共济失调评定量表; ICARS = 国际合作共济失调评定量表; MOI = 遗传模式; OCT = 光学相干断层扫描技术; OT =职业治疗; PT = 物理治疗; SARA = 共济失调评定量表
- 1.
- 2.
遗传咨询师,高级遗传护士
对症治疗
参见 Wolfram综合征临床管理指南,第6-12页的管理建议。
表8.
WFS1-WSSD患者的对症治疗
| 相关症状 | 治疗 | 注意事项/其他 | |
|---|---|---|---|
| 糖尿病 | 胰岛素依赖型糖尿病的常规治疗 | ||
| 视神经萎缩 | 校正屈光不正 | 评估视觉辅助。社区通过早期干预或开设学区进行视觉辅助服务。艾地苯醌和二十二碳六烯酸(DHA)不能改善视神经萎缩。 | |
| 感音神经性听力障碍 | SNHL的治疗取决于听力下降的程度。1 | 听力损失首先影响高频听力。 | |
| 移动 | 脚:合适的鞋;使用矫形器(鞋垫,夹板,支架)解决步态问题,改善平衡,缓解和/或改善压疮。步态训练;使用辅助行走装置(如手杖、助行器、带轮助行器、带座助行器、轮椅) | ||
| 日常生活活动 | 物理治疗师 | 位置转换(如,从床到轮椅,从轮椅到车);训练跌倒时如何减少受伤的风险 | |
| 职业治疗师 | 完成包括行动,洗漱,穿衣,吃饭,做饭和化妆等内容;对住房的格局进行改变以满足特殊需要 | ||
| 吞咽困难 | 确定吞咽困难的确切原因;调整食物的种类和一致性,吞咽时头部的位置,以及锻炼来改善吞咽。注意口腔卫生和牙科护理,因为吞咽困难可能导致清除微生物的能力下降和致病微生物定植。 | ||
| 构音障碍 | 言语病理学家 | 帮助控制声音,改善言语,呼吸技巧和日常沟通。 | |
| 脑干功能障碍 | 中枢性呼吸暂停的治疗 | ||
| 幼儿发育 | 见发育迟缓/智力残疾管理问题 | ||
| 认知能力下降/智力残疾 | |||
| 精神/行为 | 根据需要,由精神科专业人员(精神病学家、心理学家、神经心理学家)进行标准治疗 | 观察性格变化。 | |
| 神经源性膀胱功能障碍 | 抗胆碱能药物;清洁间歇性自我导尿或留置导尿管;复发性尿路感染的治疗 | ||
| 肠功能障碍 | 饮食管理 | 少食多餐,增加摄入纤维饮食,增加水摄入量 | |
| 其他内分泌 | 尿崩症 | 根据每种疾病的治疗标准进行治疗 | |
| 性腺机能减退症 | |||
| 甲状腺功能减退症 | |||
| 生长迟缓 | |||
| 家庭/社区 | 确保参与适当的社会工作,将家庭与当地资源、疗养和支持联系起来。协调医务人员以管理多个亚专科预约、设备、药物和用品。 | 考虑参加适应性运动或特殊奥林匹克运动会。 | |
DM = 糖尿病; SNHL = 感音神经性听力损失
- 1.
有关治疗方案的详细信息,请参阅遗传性听力损失和耳聋概览。
发育迟缓/智力障碍管理问题
以下信息代表了美国发育迟缓/智力障碍患者的经典管理建议;标准可能因国家而异。
0-3岁. 建议由婴儿心理健康服务、特殊教育者和感觉障碍专家对职业、物理、言语和喂养进行早期干预。在美国,早期干预是一个联邦政府资助的项目,可在所有州提供针对个人治疗需求的家庭服务。
3-5岁. 在美国,推荐当地公立学区发展学前教育。在进行学前教育之前,评估确定需要的服务和治疗,并基于当下的运动、言语、社交或认知延期情况制定个性化教育计划(IEP)。早期干预计划通常有助于改善。通常在学前教育机构进行学习;对于身体状况太不稳定以至于不能参加的儿童,提供家庭服务。
所有年龄. 建议咨询儿科医生,以确保社区、州和教育机构(美国)的参与,并支持父母尽可能地提高生活质量。以下是一些需要考虑的问题:
- 个性化教育计划 (IEP) 服务:
- 个性化教育计划为符合条件的儿童提供专门的指导和相关服务。
- IEP服务将每年审查一次,以确定是否需要进行更改。
- 根据《特殊教育法》的规定,尽可能地减少孩子们在学校里受到的限制,并在适当的时候接受普通教育。
- 为支持孩子获取学位,孩子IEP团队应包括视力和听力顾问。
- 在孩子获取学位的需求范围内,IEP将提供PT, OT和言语服务。除此之外,可能还会考虑为受累的个体提供个人支持治疗。由发育儿科医生提出关于治疗类型的具体建议。
- 当孩子进入青少年时期时,应该讨论过渡计划并纳入IEP。对于接受IEP服务的学生,公立学区必须提供服务至21岁。
- 可以考虑504计划(504计划:禁止歧视残疾的美国联邦法规),以满足那些需要辅助设施的人,如前排座位、辅助技术设备、课堂文员、课间额外时间、修改作业和放大文本。
- 建议加入发育障碍管理局 (DDA)。DDA 是美国的一个公共机构,为符合条件的个人提供服务和支持。资格因州而异,但通常由诊断和/或相关的认知/适应性残疾决定。
- 收入和资源有限的家庭也有资格获得残疾儿童的补充保障收入(SSI)。
运动功能障碍
粗大运动功能障碍
- 推荐物理治疗,以最大限度地活动和减少继发骨科并发症(如挛缩、脊椎侧弯、髋关节脱位)的风险。
- 根据需要考虑使用耐用的医疗设备和辅助设备(如轮椅、助行器、浴椅、矫形器、适应性婴儿车)。
精细动作功能障碍. 对于进食、梳洗、穿衣和写作等精细运动技能困难的,建议采用职业疗法。
沟通问题. 考虑对有语言表达困难的个体进行替代沟通方式 (比如,增强和替代沟通 [AAC]) 的评估。ACC评估可以由在该领域有专业知识的言语病理学家完成。评估将考虑认知能力和感觉障碍,以确定最合适的沟通形式。ACC设备可以是技术含量低的,如图像交换通信,也可以是技术含量高的,如语音生成设备。与人们普遍的想法相反,ACC设备并不会阻碍语言的发展,而是在很多情况下还能提高语言能力。
社会/行为的担忧
儿童可能有资格从治疗自闭症谱系病的干预措施中获益,包括应用行为分析(ABA)。ABA疗法分析个体儿童在行为、社交和适应方面的优势和劣势,通常由经过委员会认证的行为分析师一对一进行。
咨询儿科医生可能有助于指导父母调整对儿童的行为管理策略或提供处方药物,在必要时,可提供如治疗注意力缺陷/多动症(ADHD)的药物。
对于严重攻击性或破坏性行为的担忧可以咨询儿科精神病医生。
监测
参见 Wolfram综合征临床管理指南,第6-12页的监测建议。
表9.
推荐的WFS1-WSSD患者检测措施
| 相关系统 | 评估内容 | 频次 | |
|---|---|---|---|
| 糖尿病 | 参见脚注1. | 参见脚注1. | |
| 糖尿病并发症 | 肾病 | 从12岁开始每年进行一次检查 | |
| 视网膜病 | 5岁以上糖尿病患者每年筛查一次 | ||
| 神经病 | 每年检查麻木、疼痛、痉挛、感觉异常 | ||
| 血脂异常 | 参见脚注1. | ||
| 高血压 | 至少每年检查一次 | ||
| 视神经萎缩 | 视力检查(视力、色觉、白内障裂隙灯检查、眼底镜检查、视野检查);需要低视力的辅助设备 | 每年 | |
| 感音神经性听力障碍 | 听力图以及言语识别率 | 每1-2年 | |
| 神经病学 | 神经系统检查包括小脑共济失调、记忆、性格改变的评估 | 每1-2年 | |
| 日常生活活动和行动能力 | 体格检查、运动能力的OT/PT评估,自助技能 | 每次治疗时 | |
| 吞咽困难 | 评估吞咽能力 | 每次治疗时 | |
| 构音障碍 | 言语病理学家 | 每位言语病理学家 | |
| 幼儿发育 | 监督发育情况和教育需求 | 每年 | |
| 认知能力下降/智力残疾 | 每次治疗时 | 每年 | |
| 精神病学/行为学 | 评估抑郁症的迹象,自杀行为,个人外观的变化和社会行为 | 每次治疗时 | |
| 神经源性膀胱功能障碍 | 尿动力学检查和评估膀胱排空。有膀胱功能障碍或其他尿路异常时进行常规尿培养。 | 每年 | |
| 其他内分泌 | 尿崩症 | 评估尿液集中能力。 | 每次治疗时 |
| 性腺机能减退症 | 监测青春期开始的迹象。 | 每次治疗时 | |
| 甲状腺功能减退症 | 使用标准生长图表检测儿童的生长曲线。 | 每次治疗时 | |
| 生长迟缓 | 每次治疗时 | ||
| 家庭/社区 | 确保参与适当的社会工作,将家庭与当地资源、休息和支持联系起来。协调医务人员以管理多个亚专科预约、设备、药物和用品。 | ||
OT = 职业疗法; PT = 物理疗法
- 1.
详细信息请参见Wolfram综合征临床管理指南,第6-12页。
亲属风险评估
应该明确先证者无明显症状的兄弟姐妹的基因型,以便尽早确定哪些人将可能出现WFS1-WSSD的早期表现:糖尿病、视神经萎缩和感音神经性听力损失。
孕期管理
与没有糖尿病的孕妇相比,患有胰岛素依赖型糖尿病的孕妇生出先天缺陷(胚胎期糖尿病)的风险要高2-8倍。这些缺陷可累及颅面、心血管、胃肠、泌尿生殖、肌肉骨骼和中枢神经系统。对妊娠前和妊娠期间的血糖进行控制,可以减少胚胎期糖尿病的发生风险,但不能完全消除。高分辨率胎儿超声检查和胎儿超声心电图可用于筛查妊娠期间的先天的异常,还应考虑在怀孕期间咨询产科专家。
由于WFS1-WSSD患者在妊娠期间可能发生尿崩症 [Rugolo et al 2002],因此有必要在妊娠期间关注尿崩症的发生。
有关怀孕期间药物使用的进一步信息,请参阅MotherToBab。
潜在治疗方法
Abreu & Urano [2019]和Pallotta et al [2019]小组探索了新的治疗WFS1-WSSD的潜在方案。
在美国ClinicalTrials.gov和欧洲EU Clinical Trials Register这两个网站上可获取各种疾病和病症的临床实验信息。
遗传咨询
遗传咨询是向个人和家庭提供有关遗传病的病程、遗传和影响的信息,以帮助他们做出明智的医疗和个人决定的过程。接下来这一节讨论遗传风险的评估,以及利用家族史和基因检测来确定家庭成院的遗传状况。本节的目的不是解决个人可能面临的全部个体、文化的或道德的问题,或代替遗传学专家的咨询。 —ED.
携带者检测
对高危亲属进行携带者检测,需要先明确该家庭中WFS1的致病性变异。
产前检测和胚胎植入前检测
只有在受累的家庭成员中鉴定了WFS1致病性变异,才有可能进行产前诊断和胚胎植入前基因检测。
医学专业人员和家庭成员对产前检测可能存在不同的看法,特别是如果考虑到产前检测是为了终止妊娠而不是为了早期诊断。虽然大多数产前诊断中心认为产前检测是个人选择,但恰当的讨论这些问题有助于做出决定。
资源
GeneReviews的工作人员选择了以下特定疾病和/或支持组织和/或登记处,以使该疾病患者及其家人受益。GeneReviews不对其他组织提供的信息负责。有关选择标准的信息请点击这里。
- Association Syndrome de WolframMdm Nolwen Le Flochrésidence GauguinGrand-Champ 56390FrancePhone: 09 63 07 32 22
- National Library of Medicine Genetics Home Reference
- Wolfram Syndrome UKWS Support UK9 Church WayWorthing West Sussex BN13 1HDUnited KingdomPhone: 01903 211358
- Alexander Graham Bell Association for the Deaf and Hard of Hearing3417 Volta Place NorthwestWashington DC 20007Phone: 866-337-5220 (toll-free); 202-337-5220; 202-337-5221 (TTY)Fax: 202-337-8314Email: info@agbell.org
- American Diabetes Association (ADA)Phone: 1-800-DIABETES (800-342-2383)Email: AskADA@diabetes.org
- American Society for Deaf Children (ASDC)800 Florida Avenue NortheastSuite 2047Washington DC 20002-3695Phone: 800-942-2732 (Toll-free Parent Hotline); 866-895-4206 (toll free voice/TTY)Fax: 410-795-0965Email: info@deafchildren.org; asdc@deafchildren.org
- Diabetes UKUnited KingdomPhone: 0345 123 2399Fax: 020 7424 1001Email: info@diabetes.org.uk
- International Foundation for Optic Nerve Disease (IFOND)PO Box 777Cornwall NY 12518Phone: 845-534-7250Fax: 845-534-7250Email: ifond@aol.com
- National Association of the Deaf (NAD)8630 Fenton StreetSuite 820Silver Spring MD 20910Phone: 301-587-1788; 301-587-1789 (TTY)Fax: 301-587-1791Email: nad.info@nad.org
- National Federation of the Blind (NFB)200 East Wells Street(at Jernigan Place)Baltimore MD 21230Phone: 410-659-9314Fax: 410-685-5653Email: pmaurer@nfb.org
- EURO-WABB Project Registry欧盟罕见病登记为Wolfram综合征, Alström综合征和Bardet-Biedl综合征 (见 Farmer et al [2013])
- Wolfram Syndrome International Registry and Clinical StudyPhone: 314-362-8683Email: urano@wustl.edu
分子遗传学
分子遗传学和OMIM表格中的信息可能与GeneReview中其他部分的信息存在差异: 表格中可能提供了更新的信息. —ED.
表A.
WFS1 Wolfram综合征谱系障碍:基因和数据库
| 基因 | 染色体座位 | 蛋白质 | 位点特异性数据库 | HGMD | ClinVar |
|---|---|---|---|---|---|
| WFS1 | 4p16 | Wolframin | Hereditary Hearing Loss Homepage (WFS1) CCHMC - Human Genetics Mutation Database (WFS1) | WFS1 | WFS1 |
Table B.
WFS1 Wolfram综合征谱系障碍的OMIM链接 (在OMIM中查看所有)
分子机制
WFS1 Wolfram综合征谱系障碍 (WFS1-WSSD) 被认为是一种内质网(ER) 疾病。WFS1 编码wolframin 1,一种对内糖苷酶高度敏感的内质网跨膜糖蛋白 [Yamamoto et al 2006]。 Wolframin广泛表达,包括在猴子的视网膜神经节细胞和视神经胶质细胞中表达 [Yamamoto et al 2006, Luuk et al 2008]。WFS1 蛋白与其他已知蛋白缺乏同源性。
wolframin 1的确切功能尚未确定,但人们认为wolframin 1的缺乏会导致内质网应激,损害细胞周期进程,并影响钙稳态 [Zatyka et al 2008, Urano 2016, Abreu & Urano 2019]。Wolframin 1与CISD2编码的内质网膜间小蛋白无相互作用,CISD2基因的致病变异可导致WFS2 [Amr et al 2007] (见图1)。
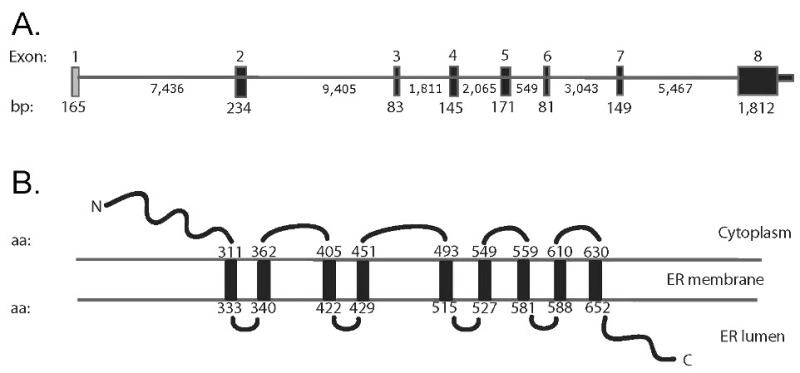
图1
A. WFS1结构示意图,由8个外显子组成,其中外显子1是不编码的。B. wolframin蛋白的预测结构。根据SMART数据库(smart.embl-heidelberg.de)预测跨膜区的位置。
致病分子机制. WFS1致病性变异导致wolframin 1蛋白功能失活。
参考文献
引用文献
- Abreu D, Urano F. Current landscape of treatments for Wolfram syndrome. Trends Pharmacol Sci. 2019;40:711 - 4. [PubMed: 31420094]
- al-Sheyyab M, Jarrah N, Younis E, Shennak MM, Hadidi A, Awidi A, El-Shanti H, Ajlouni K. Bleeding tendency in Wolfram syndrome: a newly identified feature with phenotype genotype correlation. Eur J Pediatr. 2001;160:243 - 6. [PubMed: 11317648]
- Amr S, Heisey C, Zhang M, Shows KH, Ajlouni K, Pandya A, Satin LS, El-Shanti H, Shiang R. A homozygous mutation in a novel Zinc-finger protein, ERIS, is responsible for Wolfram syndrome 2. Am J Hum Genet. 2007;81:673 - 83. [PMC free article: PMC2227919] [PubMed: 17846994]
- Barera G, Bonfanti R, Viscardi M, Bazzigaluppi E, Calori G, Meschi F, Bianchi C, Chiumello G. Occurrence of celiac disease after onset of type 1 diabetes: A 6-year prospective longitudinal study. Pediatrics. 2002;109:833 - 8. [PubMed: 11986443]
- Barrett TG. Differential diagnosis of type 1 diabetes: which genetic syndromes need to be considered? Pediatr Diabetes. 2007;8 Suppl 6:15 - 23. [PubMed: 17727381]
- Barrett TG, Bundey SE, Macleod AF. Neurodegeneration and diabetes: UK nationwide study of Wolfram (DIDMOAD) syndrome. Lancet. 1995;346:1458 - 63. [PubMed: 7490992]
- Berry V, Gregory-Evans C, Emmett W, Waseem N, Raby J, Prescott D, Moore AT, Bhattacharya SS. Wolfram gene (WFS1) mutation causes autosomal dominant congenital nuclear cataract in humans. Eur J Hum Genet. 2013;21:1356 - 60. [PMC free article: PMC3831071] [PubMed: 23531866]
- Bonnycastle LL, Chines PS, Hara T, Huyghe JR, Swift AJ, Heikinheimo P, Mahadevan J, Peltonen S, Huopio H, Nuutila P, Narisu N, Goldfeder RL, Stitzel ML, Lu S, Boehnke M, Urano F, Collins FS, Laakso M. Autosomal dominant diabetes arising from a Wolfram syndrome 1 mutation. Diabetes. 2013;62:3943 - 50. [PMC free article: PMC3806620] [PubMed: 23903355]
- Bürk K, Sival DA. Scales for the clinical evaluation of cerebellar disorders. Handb Clin Neurol. 2018;154:329 - 39. [PubMed: 29903450]
- Cano A, Molines L, Valéro R, Simonin G, Paquis-Flucklinger V, Vialettes B, et al. Microvascular diabetes complications in Wolfram syndrome (diabetes insipidus, diabetes mellitus, optic atrophy, and deafness [DIDMOAD]: an age- and duration-matched comparison with common type 1 diabetes. Diabetes Care. 2007a;30:2327 - 30. [PubMed: 17536072]
- Cano A, Rouzier C, Monnot B, Conrath J, Lecomte P, Delobel B, Boileau P, Valero R, Procaccio V, Paquis-Flucklinger V, et al. Identification of novel mutations in WFS1 and genotype-phenotype correlation in Wolfram syndrome. Am J Med Genet A. 2007b;143A:1605 - 12. [PubMed: 17568405]
- Chaussenot A, Rouzier C, Quere M, Plutino M, Ait-El-Mkadem S, Bannwarth S, Barth M, Dollfus H, Charles P, Nicolino M, Chabrol B, Vialettes B, Paquis-Flucklinger V. Mutation update and uncommon phenotypes in a French cohort of 96 patients with WFS1-related disorders. Clin Genet. 2015;87:430 - 9. [PubMed: 24890733]
- Colosimo A, Guida V, Rigoli L, Di Bella C, De Luca A, Briuglia S, Stuppia L, Salpietro DC, Dallapiccola B. Molecular detection of novel WFS1 mutations in patients with wolfram syndrome by a DHPLC-based assay. Hum Mutat. 2003;21:622 - 9. [PubMed: 12754709]
- Cryns K, Sivakumaran TA, Van den Ouweland JMW, Pennings RJE, Cremers CWRJ, Flothmann K, Young T-L, Smith RJH, Lesperance MM, Van Camp G. Mutational spectrum of the WFS1 gene in Wolfram syndrome, nonsyndromic hearing impairment, diabetes mellitus, and psychiatric disease. Hum Mutat. 2003;22:275 - 87. [PubMed: 12955714]
- Dhalla MS, Desai UR, Zuckerbrod DS. Pigmentary maculopathy in a patient with Wolfram syndrome. Can J Ophthalmol. 2006;41:38 - 40. [PubMed: 16462870]
- Domènech E, Gómez-Zaera M, Nunes V. WFS1 mutations in Spanish patients with diabetes mellitus and deafness. Eur J Hum Genet. 2002;10:421 - 6. [PubMed: 12107816]
- Domenech E, Gomez-Zaera M, Nunes V. Wolfram/DIDMOAD syndrome, a heterogenic and molecularly neurodegenerative disease. Pediatr Endocrinol Rev. 2006;3:249 - 57. [PubMed: 16639390]
- Eiberg H, Hansen L, Kjer B, Hansen T, Pedersen O, Bille M, Rosenberg T, Tranebjærg L. Autosomal dominant optic atrophy associated with hearing impairment and impaired glucose regulation caused by a missense mutation in the WFS1 gene. J Med Genet. 2006;43:435 - 40. [PMC free article: PMC2649014] [PubMed: 16648378]
- Elli FM, Ghirardello S, Giavoli C, Gangi S, Dioni L, Crippa M, Finelli P, Bergamaschi S, Mosca F, Spada A, Beck-Peccoz P. A new structural rearrangement associated to Wolfram syndrome in a child with a partial phenotype. Gene. 2012;509:168 - 72. [PubMed: 22771918]
- El-Shanti H, Lidal AC, Jarrah N, Druhan L, Ajlouni K. Homozygosity mapping identifies an additional locus for Wolfram syndrome on chromosome 4q. Am J Hum Genet. 2000;66:1229 - 36. [PMC free article: PMC1288190] [PubMed: 10739754]
- Genís D, Dávalos A, Molins A, Ferrer I. Wolfram syndrome: a neuropathological study. Acta Neuropathol. 1997;93:426 - 9. [PubMed: 9113209]
- Giuliano F, Bannwarth S, Monnot S, Cano A, Chabrol B, Vialettes B, Delobel B, et al. Wolfram syndrome in French population: Characterization of novel mutations and polymorphisms in the WFS1 gene. Hum Mutat. 2005;25:99 - 100. [PubMed: 15605410]
- Grenier J, Meunier I, Daien V, Baudoin C, Halloy F, Bocquet B, Blanchet C, Delettre C, Esmenjaud E, Roubertie A, Lenaers G, Hamel CP. WFS1 in optic neuropathies: mutation findings in nonsyndromic optic atrophy and assessment of clinical severity. Ophthalmology. 2016;123:1989 - 98. [PubMed: 27395765]
- Hansen L, Eiberg H, Barrett T, Kjaersgaard P, Tranebjærg L, Rosenberg T. Mutation analysis of the WFS1 gene in seven Danish Wolfram syndrome families; four new mutations identified. Eur J Hum Genet. 2005;13:1275 - 84. [PubMed: 16151413]
- Hardy C, Khanim F, Torres R, Scott-Brown M, Seller A, Poulton J, Collier D, Kirk J, Polymeropoulos M, Latif F, Barrett T. Clinical and molecular genetic analysis of 19 Wolfram syndrome kindreds demonstrating a wide spectrum of mutations in WFS1. Am J Hum Genet. 1999;65:1279 - 90. [PMC free article: PMC1288280] [PubMed: 10521293]
- Ito S, Sakakibara R, Hattori T. Wolfram syndrome presenting marked brain MR imaging abnormalities with few neurologic abnormalities. Am J Neuroradiol. 2007;28:305 - 6. [PubMed: 17297000]
- Jónsson H, Sulem P, Kehr B, Kristmundsdottir S, Zink F, Hjartarson E, Hardarson MT, Hjorleifsson KE, Eggertsson HP, Gudjonsson SA, Ward LD, Arnadottir GA, Helgason EA, Helgason H, Gylfason A, Jonasdottir A, Jonasdottir A, Rafnar T, Frigge M, Stacey SN, Th Magnusson O, Thorsteinsdottir U, Masson G, Kong A, Halldorsson BV, Helgason A, Gudbjartsson DF, Stefansson K. Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature. 2017;549:519 - 22. [PubMed: 28959963]
- Khanim F, Kirk J, Latif F, Barrett TG. WFS1/Wolframin mutations, Wolfram syndrome, and associated diseases. Hum Mutat. 2001;17:357 - 67. [PubMed: 11317350]
- Liu Z, Sakakibara R, Uchiyam T, Yamamoto T, Ito T, Ito S, Awa Y, Odaka T, Yamaguchi T, Hattori T. Bowel dysfunction in Wolfram syndrome. Diabetes Care. 2006;29:472 - 3. [PubMed: 16443921]
- Lusk L, Black E, Vengoechea J. Segregation of two variants suggests the presence of autosomal dominant and recessive forms of WFS1-related disease within the same family: expanding the phenotypic spectrum of Wolfram syndrome. J Med Genet. 2020;57:121 - 3. [PubMed: 31363008]
- Luuk H, Koks S, Plaas M, Hannibal J, Rehfeld JF, Vasar E. Distribution of Wfs1 protein in the central nervous system of the mouse and its relation to clinical symptoms of the Wolfram syndrome. J Comp Neurol. 2008;509:642 - 60. [PubMed: 18551525]
- Mozzillo E, Delvecchio M, Carella M, Grandone E, Palumbo P, Salina A, Aloi C, Buono P, Izzo A, D'Annunzio G, Vecchio G, Orrico A, Genesio R, Simonelli F, Franzese A. A novel CISD2 intragenic deletion, optic neuropathy and platelet aggregation defect in Wolfram syndrome type 2. BMC Med Genet. 2014;15:88. [PMC free article: PMC4121299] [PubMed: 25056293]
- Pakdemirli E, Karabulut N, Bir LS, Sermez Y. Cranial magnetic resonance imaging of Wolfram (DIDMOAD) syndrome. Australas Radiol. 2005;49:189 - 91. [PubMed: 15845065]
- Pallotta MT, Tascini G, Crispoldi R, Orabona C, Mondanelli G, Grohmann U, Esposito S (2019). Wolfram syndrome, a rare neurodegenerative disease: from pathogenesis to future treatment perspectives J Tranl Med 17: 238. [PMC free article: PMC6651977] [PubMed: 31337416]
- Pennings RJ, Huygen PL, van den Ouweland JM, Cryns K, Dikkeschei LD, Van Camo G, Cremers CW. Sex-related hearing impairment in Wolfram syndrome patients identified by inactivating WFS1 mutations. Audiol Neurootol. 2004;9:51 - 62. [PubMed: 14676474]
- Plantinga RF, Pennings RJ, Huygen PL, Bruno R, Eller P, Barrett TG, Vialettes B, Paquis-Fluklinger V, Lombardo F, Cremers CW. Hearing impairment in genotyped Wolfram syndrome patients. Ann Otol Rhinol Laryngol. 2008;117:494 - 500. [PubMed: 18700423]
- Ristow M. Neurodegenerative disorders associated with diabetes mellitus. J Mol Med. 2004;82:510 - 29. [PubMed: 15175861]
- Rondinelli M, Novara F, Calcaterra V, Zuffardi O, Genovese S. Wolfram syndrome 2: a novel CISD2 mutation identified in Italian siblings. Acta Diabetol. 2015;52:175 - 8. [PubMed: 25371195]
- Rouzier C, Moore D, Delorme C, Lacas-Gervais S, Ait-El-Mkadem S, Fragaki K, Burté F, Serre V, Bannwarth S, Chaussenot A, Catala M, Yu-Wai-Man P, Paquis-Flucklinger V. A novel CISD2 mutation associated with a classical Wolfram syndrome phenotype alters Ca2+ homeostasis and ER-mitochondria interactions. Hum Mol Genet. 2017;26:1599 - 1611. [PMC free article: PMC5411739] [PubMed: 28335035]
- Rugolo S, Mirabella D, Palumbo MA, Chiantello R, Fiore G. Complete Wolfram's syndrome and successful pregnancy. Eur J Obstet Gynecol Reprod Biol. 2002;105:192 - 3. [PubMed: 12381487]
- Shannon P, Becker L, Deck J. Evidence of widespread axonal pathology in Wolfram syndrome. Acta Neuropathol. 1999;98:304 - 8. [PubMed: 10483789]
- Simsek E, Simsek T, Hosal S, Seyrantepe V, Aktan G. Wolfram syndrome (DIDMOAD) syndrome: a multidiscliplinary clinical study in nine Turkish patients and review of the literature. Acta Paediatr. 2003;92:55 - 61. [PubMed: 12650300]
- Skovbjerg H, Tarnov L, Locht H, Parving HH. The prevalence of coeliac disease in adult Danish patients with type 1 diabetes with ad without nephropathy. Diabetologia. 2005;48:1416 - 7. [PubMed: 15918021]
- Smith CJA, Crock PA, King BR, Meldrum CJ, Scott RJ. Phenotype-genotype correlations in a series of Wolfram syndrome families. Diabetes Care. 2004;27:2003 - 9. [PubMed: 15277431]
- Stenson PD, Mort M, Ball EV, Evans K, Hayden M, Heywood S, Hussain M, Phillips AD, Cooper DN. The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet. 2017;136:665 - 77. [PMC free article: PMC5429360] [PubMed: 28349240]
- Swift RG, Polymeropoulos MH, Torres R, Swift M. Predisposition of Wolfram syndrome heterozygotes to psychiatric illness. Mol Psychiatry. 1998;3:86 - 91. [PubMed: 9491819]
- Tessa A, Carbone I, Matteoli MC, Bruno C, Patrono C, Patera IP, De Luca F, Lorini R, Santorelli FM. Identification of novel WFS1 mutations in Italian children with Wolfram syndrome. Hum Mutat. 2001;17:348 - 9. [PubMed: 11295831]
- Tranebjærg L. Wolframin 1-related disease and hearing. In: Kóks S, Vasar E, eds. Emerging Link Between the Emotional Brain and Endocrine Pancreas. Kerala, India: Research Signpost; 2008:107-24.
- Urano F. Wolfram syndrome: diagnosis, management, and treatment. Curr Diab Rep. 2016;16:6. [PMC free article: PMC4705145] [PubMed: 26742931]
- Valéro R, Bannwarth S, Roman S, Paquis-Flucklinger V, Vialettes B. Autosomal dominant transmission of diabetes and congenital hearing impairment secondary to a missense mutation in the WFS1 gene. Diabet Med. 2008;25:657 - 61. [PubMed: 18544103]
- van den Ouweland JM, Cryns K, Pennings RJE, Walraven I, Janssen GMC, Maassen JA, Veldhuijzen BFE, Arntzeniue AB, Lindhout D, Cremers CWRJ, Van Camp G, Dikkeschei LD. Molecular characterization of WFS1 in patients with Wolfram syndrome. J Mol Diagn. 2003;5:88 - 95. [PMC free article: PMC1907324] [PubMed: 12707373]
- Yamamoto H, Hofman S, Hamasaki DI, Yamomoto H, Kreczmanski P, Schmitz C, Parel J-M, Schmidt-Kastner R. Wolfram syndrome 1 (WFS1) protein expression in retinal ganglion cells and optic nerve glia of the cynomolgus monkey. Exp Eye Res. 2006;83:1303 - 6. [PubMed: 16928372]
- Zatyka M, Ricketts C, Xavier GS, Minton J, Fenton S, Hofman-Thiel S, Rutter GA, Barrett TG. Sodium-potassium ATPase β1 subunit is a molecular partner of Wolframin, and endoplasmic reticulum protein involved in ER stress. Hum Mol Genet. 2008;17:190 - 200. [PMC free article: PMC6101208] [PubMed: 17947299]
说明
致谢
由Lisbeth Tranebjærg领导的听力遗传学研究小组得到了Widex AS和其他研究基金的财政支持。
修订史
- 2020年4月9日 (bp) 全面更新
- 2013年12月19日 (me) 全面更新
- 2009年6月2日 (cd) 修订: 缺失/重复分析可以用于临床
- 2009年2月24日 (me) 综述发布
- 2008年8月12日 (lt) 原始提交版