
摘要
临床特征.
Wolf-Hirschhorn 综合征(WHS)婴儿时期的典型颅面部特征为“希腊战士钢盔样”鼻(宽鼻梁延伸到前额)、 小头畸形、前发际高、眉间突出、眼距宽、内眦赘皮、高拱形眉毛、人中短、嘴角下弯、小下颌畸形及耳朵有凹陷或赘生物。所有 患病个体表现为产前生长缺陷、产后发育迟缓以及肌肉发育不良导致的肌张力低下。所有患者都有不同程度发育迟缓和智力障碍。90%-100%的WHS患儿发生癫痫。 其他临床表现包括骨骼异常(60% -70%)、先天性心脏缺陷(50% )、听力丧失(多数为传导性耳聋)(>40%)、泌尿系统畸形 (25% )和脑结构异常(33% )。
诊断/测试.
采用染色体微阵列分析(CMA)、传统G - 显带细胞遗传学分析或荧光原位杂交技术( FISH)在 4号染色体短臂1区6带第三亚带(4p16.3 )WHS关键区域(WHSCR)发现杂合缺失即可诊断为WHS 。
治疗.
对症治疗: 疗法包括: 康复治疗、 语言/沟通治疗和手语;丙戊酸治疗非典型性癫痫发作;苯二氮平类药物治疗癫痫持续状态;针对喂养困难可采用特殊的喂养技术,如填胃法或胃造口术.
针对骨骼畸形、眼部异常、 先天性心脏缺陷、听力丧失、睡眠障碍及肝腺瘤,推荐采用标准治疗。并发症预防:抗生素预防膀胱输尿管反流;免疫球蛋白注射或持续性抗生素治疗抗体缺乏患者。
监测:按需进行系统的随访以监控康复和治疗;每年进行全血细胞计数和肾功能检测;常规肝脏超声波检查
应避免的因子/环境: 卡马西平可能会加重非典型性癫痫发作。.
遗传咨询.
导致WHS的原因是多种不同机制引起的4p16.3染色体WHSCR区域缺失。约50%-60% 的WHS个体有一个4p16新发纯合缺失,约40%-45%的患者有一个4号染色体短臂缺失和另一染色体臂部分三体的非平衡 易位。 这些非平衡易位可能是新发的,也可能是遗传自父亲或母亲的平衡重排。余下的 4p16.3 缺失由其他复杂的染色体重排导致(例如,4号环状染色体)。家庭成员的风险取决于缺失的产生机制。若双亲之一是 4p16.3相关染色体重排 携带者,应考虑产前检查。
诊断
推测性结论
有以下临床表现的个体应考虑为WHS:
典型面容 [Battaglia et al 1999a, Battaglia et al 1999b, Battaglia et al 2000, Battaglia & Carey 2000, Battaglia et al 2008] (see Figure 1):
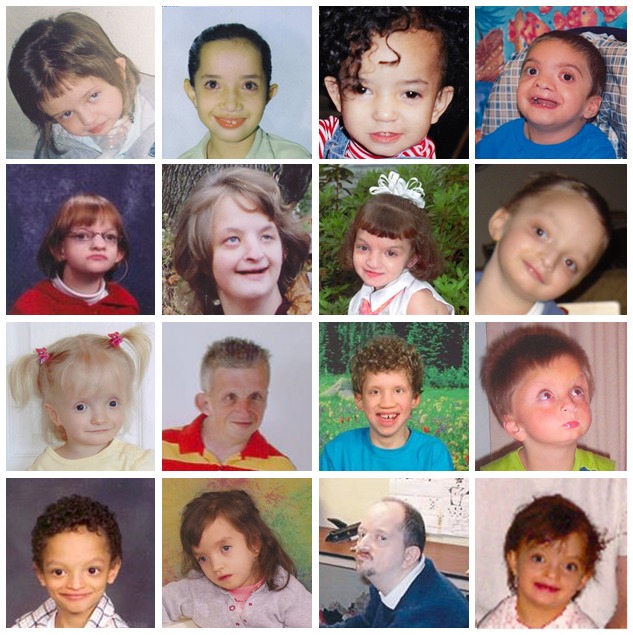
图 1.
16个不同年龄WHS个体的面部外观,转载自 South等 [2008c]
- “希腊战士钢盔样”鼻 (宽鼻梁延伸到前额)
- 小头畸形
- 前发际高、眉间突出
- 眼距宽
- 内眦赘皮
- 高拱形眉毛
- 人中短
- 嘴角下弯
- 小下颌畸形
- 耳朵有凹陷/赘生物
产前生长缺陷,产后发育迟缓
不同程度发育迟缓/智力障碍
癫痫和/或特殊的脑电图异常 [Battaglia et al 2009]
肌张力减退和肌肉发育不良,主要在下肢
确诊
WHS的诊断依据是,在先证者的4p16.3WHS关键区域(WHSCR) 内检测到一个1.4~1.9兆的杂合缺失(详见表 1)。4p16.3WHSCR缺失的大致位置是参考基因组的chr4: 419,224-2,010,962 (NCBI BuildGRCh37/hg19)。
注: 该区域内显著更大或更小的缺失,它的临床表型可能不同于WHS (详见遗传相关疾病)。
确定序列拷贝数的 遗传学检测方法包括染色体微阵列分析(CMA)、传统G-显带 细胞遗传学分析和用荧光原位杂交技术(FISH)进行靶向缺失分析。表 1.
使用基因组测试检测WHS
高危亲属风险评估.采用FISH检测 先证者高危亲属是否在4p16.3WHSCR内存在缺失。父母样本的测定对再发风险的评估很重要(详见遗传咨询).
检测特点. 关于检测特点中的灵敏度和特异性,详见 Clinical Utility Gene Card [Battaglia et al 2011] .
临床特点
临床描述
典型 WHS. 表2 总结了WHS相关临床表现的频率分布.
表 2.
WHS综合征相关临床表现的频率分布
| 临床表现 1 | 频率 |
|---|---|
典型的面部特征 (详见 Suggestive Findings) | >75% |
皮肤改变 (血管瘤; 大理石样皮肤/干皮) | 50%-75% |
听力损伤 刻板(洗手/拍打、摆动) | 25%-50% |
以下异常:
| <25% |
面部特征. 从出生到孩童时期,患者鼻子外观似“希腊战士钢盔样” (宽鼻梁延伸到前额),在青春期时变得不明显。
产后发育迟缓. 大多数WHS个体都有明显的宫内发育迟缓和身材矮小,尽管后天摄入了足够的能量和蛋白质,体重仍然增长缓慢[ Battaglia et al 1999a, Battaglia et al 1999b, Battaglia & Carey 2000, Battaglia et al 2008]。该类患者从出生到4岁的特异性生长曲线已被绘制 [Antonius et al 2008]. 除了有隐秘非平衡易位的个体,其他所有 患病个体的头围小于第二个百分位数[South et al 2008c]。
智力障碍. 虽然人们普遍认为WHS个体具有严重的智力障碍,无语言能力,沟通能力差,但是在WHS个体中观察到广泛范围的智力。 Battaglia 等 [2008]发现智力障碍程度轻度的约10%、 中度约25%、 重度约65%。因此,三分之一 患病个体有轻度到中度的失能。大多数WHS个体的语言表达能力仅限于喉音和双音节词,6%的个体能够表达简单的句子。理解能力似乎只限于特定的环境中。 大多数WHS患者尝试去沟通,随着时间的推移和手势的拓展,他们的沟通能力得到提高。Fisch 等[2010]研究了19个具有表达能力和语言能力的WHS 儿童,观察他们社会化 范围内的相对优势。
约10% 患病个体具有括约肌控制能力,多数在8-14岁。在两岁到12岁的WHS儿童中,接近45%的患者可以行走,其中25%可以独立行走,20%可以在物体的支持下行走 [Battaglia & Carey 2000, Battaglia et al 2008]。大约 30% 的儿童生活具有自主性(10%能够自主进食,20%能够自主穿衣和脱衣),能够做简单的家务。随着时间的推移,所有WHS个体都有缓慢但持续的改善; 这些个体各方面的改善超越了之前的预期。
癫痫.90%-100%的WHS儿童有癫痫发作[Battaglia et al 1999a, Battaglia et al 1999b, Battaglia & Carey 2000, Battaglia et al 2009]。发病年龄为3~23个月,发病高峰在6~12个月。 癫痫通常表现为单侧阵挛或强直,伴或不伴继发性全身性发作,或从发病开始即为全身强直性阵挛。癫痫通常由发热引起,痉挛成簇出现且持续时间大于15分钟。
其它癫痫发作类型包括强直性痉挛、肌阵挛性癫痫发作和复杂的部分性癫痫发作 [Battaglia & Carey 2005]。多达50%的患者出现癫痫持续状态。三分之一的WHS儿童在1-6岁时出现非典型性癫痫发作[ Battaglia et al 2009]。
某些患者的癫痫发作早期,症状很难控制。但是适当治疗后,癫痫会随着年龄的增长而消失。多达55%的患者在两岁到13岁时,癫痫停止发作[ Battaglia et al 2009]。
90%WHS个体出现独特的脑电图异常(EEG), 包括由睡眠激活的发生在长波区的2-3.5赫兹的弥散性不明确的尖棘峰/复合波;由闭眼触发的,在头部的后三分之一处的4-6赫兹的高幅度尖棘峰/复合波。[ Battaglia et al 2009].
喂养困难可能由肌张力低下或面裂相关的吮吸困难、吞咽和吮吸不协调、或胃食管反流导致。胃食管反流,在正常婴儿中短暂出现,在WHS婴儿中通常持续出现,最终导致WHS婴儿生长发育不良和呼吸道疾病。
骨骼异常. 60%-70%的WHS个体出现骨骼异常[Battaglia et al 1999a, Battaglia et al 1999b, Battaglia & Carey 2000, Battaglia et al 2008] , 包括脊柱后凸畸形(驼背)/脊柱侧弯、附件或肋骨融合、畸形足和手劈裂[ Shanske et al 2010]。
眼睛异常. WHS患者最常见的眼部表现有外隐斜视、鼻泪管阻塞、眼睛或视神经缺损和中央凹发育不良[ Battaglia et al 2001, Wu-Chen et al 2004, Battaglia et al 2008].偶可观察到眼睑发育不良,需要皮肤移植 [ Battaglia et al 2001]。青光眼很难治疗。
牙齿发育异常. 大于50%的患者出现牙齿发育不全,表现为牙齿萌出延迟,牙齿长期以乳牙形式存在,如,长冠牙和钉型牙[ Battaglia & Carey 2000, Battaglia et al 2001, Battaglia et al 2008].
约50% 的患者出现先天性心脏缺陷,通常并不复杂。最常见的是房间隔缺损(27%), 其次是肺动脉瓣狭窄、室间隔缺损、动脉导管未闭、主动脉瓣关闭不全和法洛四联症 [Battaglia et al 1999a, Battaglia et al 1999b, Battaglia & Carey 2000, Battaglia et al 2008].
抗体缺陷.69%的WHS儿童出现IgA/IgG2亚类缺陷、 单纯性IgA 缺乏综合征或受损的多糖反应Hanley-Lopez et al [1998] ,这可能是导致反复呼吸道感染和中耳炎的原因。
听力丧失. 大于40%的WHS个体出现听力丧失,多为传导性听力障碍。 约15%的个体出现感音神经性耳聋 [Battaglia & Carey 2000, Battaglia et al 2008]. 先天性中耳和内耳异常可能是导致听力损害的原因 [Ulualp et al 2004].
尿路畸形在超过30%的患病个体中出现,包括肾发育不全、囊性发育不良/发育不全,寡巨肾小球病(特点为肾单位数目减少和所有 肾部肥大的肾发育不全)(马蹄肾、肾旋转不良、膀胱外翻和尿路梗阻。寡巨肾小球病与慢性肾功能衰竭有关。其中一些异常与膀胱输尿管反流有关 [Battaglia & Carey 2000, Grisaru et al 2000, Battaglia et al 2008].
50%的男性出现尿道下裂和隐睾[Battaglia & Carey 2000].
女性表现为无子宫、生殖腺退化和阴蒂发育不全/增生[Battaglia et al 2008].
中枢神经系统结构异常.多达80%的患病个体出现中枢神经系统结构异常[ Battaglia et al 2008]。异常主要包括胼胝体变薄,少数病例有白质体积弥散性减少、侧脑室增宽、皮质/皮质下萎缩和双侧小脑半球发育不良。 其他报道的大脑发育不全包括脑回狭窄、无嗅脑、海马H2区缩短和小脑内脑回异位发育不良[ Battaglia & Carey 2000].
睡眠问题常发生在早期,如不是由中耳炎、胃食管反流、湿疹和阻塞性睡眠呼吸暂停综合症等临床原因引起,则较易治疗[ Battaglia et al 2001]。
其他. 少数WHS个体的许多先天性异常也有报道 [Battaglia et al 2001].
- 造血功能障碍. 据报道,有两例WHS儿童出现造血功能障碍:一例由造血功能障碍发展成难治性贫血,另一例发展为急性淋巴细胞白血病 [Sharathkumar et al 2003].
- 肝腺瘤. 据报道,有3例WHS个体患有肝腺瘤,其中一例发展为肝癌 [Calhoun et al 2013, Prunotto et al 2013]. 需要进一步深入研究来更好的阐明这些WHS并发症的发生机制。
基因型-表型 相关性
为了解释WHS的表型多样性,研究人员一直在探究4p 缺失大小与临床表现严重程度之间的相关性。
一些研究者提出基因-表型相关性部分或完全相关 [Wieczorek et al 2000, Zollino et al 2000, Zollino et al 2008], 另一些研究者认为无相关性存在 [Battaglia et al 1999a, Battaglia et al 1999b]. Meloni等[2000] 观察到无论是具有严重智力障碍的典型WHS患者,还是具有轻、中度智力障碍的WHS患者,采用常规 细胞遗传学检查没有发现大片段畸形,采用 FISH检查发现了亚微观缺失。这些发现表明缺失的大小与临床严重程度无关。 一些相关的结构缺陷,如腭裂和心脏缺陷,在缺失大于3兆的个体上出现较频繁 [Zollino et al 2008].
经典的表型可能包括WHS患者少数典型异常和非平衡 易位导致的另一染色体部分三体。
It has been shown that double cryptic 染色体 imbalances。
最初被误认为是微缺失,实际上它是由一个与非平衡易位相关的较大缺失导致,是WHS表型多样性的重要因素[Zollino et al 2004]。缺失大小与表型严重性部分相关,但某些个体 患病程度比根据缺失大小预计的更严重或更轻。
遗传相关性(等位基因) 疾病
4P端长度达22~25兆的缺失与一种不同于WHS临床表现的严重表型相关[Zollino et al 2008].
WHSCR远端部分的缺失可能是良性的,也可能与轻度生长发育迟缓、癫痫发作有关,但没有WHS的诊断特征[ South et al 2008c].
鉴别诊断
4p近端 缺失. 已经描述了几个中段缺失的个体. 此类缺失通常涉及4p12-p16区域,不在WHS关键区域,而在该区域近端。该疾病是一种离散综合征,不同于WHS [Bailey et al 2010].
WHS 表型. WHS的临床表型尤其是面部形态非常典型; 然而,仍有些个体因为某些特征与以下疾病重叠而被误诊:
- Angelman syndrome(AS) 的特征是严重的发育迟滞或智力障碍,严重的语言障碍、步态共济失调/颤抖的四肢,不合时宜的愉悦,包括频繁大笑、 微笑和兴奋。常见小头畸形和癫痫。发育迟缓首次出现是在六个月左右;然而,AS个体直到一岁后才表现出独特的临床特征,并且需要几年时间才能做出正确 的临床诊断。AS 是由位于15q11.2-q13 天使综合症或普拉达威利综合症(AS/PWS) 区域内的母源性印记基因UBE3A被破坏所致。
处理
初步诊断后评估
推荐以下评估手段来确立WHS个体的疾病状况和需求:
- 生长参数的测量和生长曲线图的绘制
- 认知、语言、运动发育和社交技能的评估
- 清醒/睡眠动态多重脑电图检测儿童时期(主要年龄段是1-6岁)非典型性癫痫发作[Battaglia & Carey 2000, Battaglia et al 2009]
- 评估吞咽困难组的喂养问题和胃食管反流
- 骨骼异常体格检查(畸形足、脊柱侧弯、脊柱后凸畸形(驼背));如有异常,转诊矫形外科和物理治疗评估(包括完整的生物力学评估)
- 即使无明显异常,也应在婴儿期进行眼科会诊
- 婴儿期心脏检查(听诊、心电图、超声心动图)
- 免疫缺陷测试(特别是血浆免疫球蛋白水平、淋巴细胞亚群和多糖反应);虽然WHS个体的免疫缺陷资料有限,但在临床适用的条件下可以考虑
- 全血细胞计数评估造血功能障碍
- 尽早进行耳鼻喉和听力学筛查综合评估(脑干听觉诱发反应),以便采取适当干预措施
- 婴儿期肾功能检查和肾超声检查检测肾脏结构异常/膀胱输尿管反流 [Grisaru et al 2000]
- 基线肝脏超声检查评估肝脏腺瘤
- 咨询医学遗传学家/遗传咨询专家
对症处理
智力障碍. 个体化康复方案中应包括运动发育、认知、沟通和社交技能 [Battaglia & Carey 2000, Battaglia et al 2008]。使用手语不仅可以提高沟通技能,而且不会抑制语言的出现。早期干预和后期适当的学校安置非常必要。
癫痫. 约95%的WHS患者有多次癫痫发作,常由发热引起,近三分之一的患者随后发展为丙戊酸-反应型不典型癫痫发作,因此在癫痫首次发作后应立即使用丙戊酸治疗。单独使用丙戊酸或联合使用ethosuccimide可以很好的控制非典型癫痫发作。 [Battaglia & Carey 2000, Battaglia et al 2009].
最近提出溴化钠可作为预防癫痫持续状态的初始治疗方法 [Kagitani-Shimono et al 2005].
静脉注射苯二氮卓类(地西泮)可以很好的控制阵挛、强直阵挛、癫痫发作和肌阵挛性癫痫持续状态 [Battaglia & Carey 2005, Kagitani-Shimono et al 2005].
WHS个体独特的脑电图异常不一定与癫痫发作有关[Battaglia et al 2009],因此,在个体不再有癫痫发作5年后, 可以退出抗癫痫药物治疗。
喂养困难. 这些患者适合使用关注口腔运动技能的喂养疗法。特殊的喂养技术或设备,如“Haberman feeder”,可用于无腭裂或未手术修复腭裂的低肌张力婴儿/儿童。
灌胃强饲法适用于吞咽能力差的个体。
胃食管反流应用标准的方式处理。
在某项研究中,近44%的WHS个体采用胃造口术、偶采用胃底折叠术 [Battaglia & Carey 2000].
骨骼畸形(例如,足畸形、脊柱侧弯、驼背)根据个体需要解决。建议早期治疗(物理治疗和手术治疗)
眼部异常用标准方法处理.
先天性心脏缺陷通常不复杂,且易于修复.
听力丧失可使用助听器.
睡眠问题. 如果不是医学因素(如中耳炎、胃食管反流、湿疹、阻塞性睡眠呼吸暂停)引起,或者父母的关注加重了睡眠问题,则“父母注意力消失”是一种有效的行为疗法。[ Curfs et al 1999].
肝腺瘤.医学处理(手术治疗或化疗)的差异与腺瘤的数量和大小有关.
其他 结构异常 (如, 隔膜、肠胃系统、牙齿) 应采用标准疗法.
并发症的预防
预防性抗生素治疗膀胱输尿管反流.
静脉注射免疫球蛋白或连续使用抗生素治疗抗体缺陷.
监测
采用系统的随访进行监测,根据患者技能的改善或恶化及医疗需求进行康复和治疗方案的调整 [Ferrarini et al 2003, Battaglia et al 2008, Battaglia 2010].
- 每年全血细胞计数评估造血功能异常
- 每年进行肾功能检测,包括血清尿素氮、肌酐、胱抑素C;尿液分析;肌酐清除率试验
- 采用常规肝脏超声检查来评估肝腺瘤
应避免的因子/环境
卡马西平可能恶化非典型性癫痫发作[Battaglia & Carey 2005].
还在研究阶段的治疗方法
搜索ClinicalTrials.gov获取各种疾病的临床研究信息。注:该疾病可能没有临床试验。
遗传咨询
遗传咨询是指向患者本人及其家庭提供遗传性疾病的性质、遗传方式及其可能造成的影响等信息 ,以帮助他们做出知情的医疗及个人决定。以下章节涉及遗传风险评 估,根据家族史和遗传学检测来确定家庭成员的遗传状态。此章节目的并不是为了解决所有患者可能面临的个人、文化或伦理问题,或者企图替代遗传学专业人员的 咨询工作。-ED
相关遗传咨询问题
4号染色体短臂(4p)和其他染色体易位的具体经验风险值未知。然而,采用FISH分析父母亲4号染色体短臂(4p)亚端粒可排除这种可能性。遗传咨询适用于有再发风险的家庭。
研究表明,不同子代之间终端缺失片段的大小有差异[Faravelli et al 2007, South et al 2008b]。这在4号染色体短臂(4p)和8号染色体长臂(18q)中有所描述。
The risk for such a finding in a clinically unaffected parent is unknown at present.。
生育计划
- 怀孕前是评估遗传风险、明确携带者状态、对产前检查进行可行性分析的最佳时机。
产前检查
高风险孕妇. 在已知父亲或母亲是4号染色体短臂(4p)重排携带者的家庭,应考虑产前检测。根据先证者及其父母的特异性临床表现,通过绒毛取样(通常在妊娠第10-12周进行)或羊膜腔穿刺术(通常在妊娠第15-18周进行)获取细胞,然后联合 细胞遗传学方法进行分析(G-显带、FISH和CMA).
注:孕周以月经周数的形式表达,可从末次月经的第一天开始计算,也可通过超声测量。.
低风险孕妇. 宫内发育迟缓(IUGR)的胎儿,三维(3D)超声显示其类似希腊战士钢盔样面部特征[ Chen et al 2004].
胚胎植入前遗传学诊断(PGD). 对有可能因遗传染色体重排而有孕WHS胎儿风险的孕妇,可选择胚胎植入前遗传学诊断。
信息来源
为了帮助患者及其家庭,GeneReviews员工已经选择了针对下列疾病的患者支持组织。 GeneReviews不为其他组织提供的信息承担责任。 关于选择这些组织的标准,请点击此处.
- 4P-Support Group, Inc2159 128th StreetNew Richmond WI 54017Phone: 715-248-3937
- Associazione Italiana Sindrome di Wolf-Hirschhorn (AISiWH)Via Bologna 65Montecosaro 62010ItalyPhone: 0733 864275Fax: 0733 864275Email: segreteria.aisiwh@gmail.com
- My46 Trait Profile
- Wolf Hirschhorn Syndrome Trust (WHST)United KingdomPhone: 0845 603 5338Email: enquiries@whs4pminus.co.uk
- Chromosome Disorder Outreach (CDO)PO Box 724Boca Raton FL 33429-0724Phone: 561-395-4252 (Family Helpline)Email: info@chromodisorder.org
分子遗传学
在分子遗传学和OMIM表内的信息可能和GeneReview里其他部分不完全一致,表内信息较新. —ED.
表 A.
WHS: 基因和数据库
表B.
WHS的OMIM入口 (View All in OMIM)
| 194190 | WOLF-HIRSCHHORN SYNDROME; WHS |
| 602618 | C-TERMINAL-BINDING PROTEIN 1; CTBP1 |
| 602952 | NUCLEAR RECEPTOR-BINDING SET DOMAIN PROTEIN 2; NSD2 |
| 604407 | LEUCINE ZIPPER/EF-HAND-CONTAINING TRANSMEMBRANE PROTEIN 1; LETM1 |
| 605032 | COMPLEXIN 1; CPLX1 |
| 605830 | FIBROBLAST GROWTH FACTOR RECEPTOR-LIKE 1; FGFRL1 |
| 606026 | WHS CANDIDATE 2 GENE; WHSC2 |
分子遗传机制
通过两个WHS个体表型的确定和一个包含候选基因 LETM1和WHSC1的长度为1.9兆的末端缺失来界定WHSCR的近端边界[Zollino et al 2003, Rodríguez et al 2005]。通过分析有4p16.3中段缺失及WHS表型的个体 [Wright et al 1997] 和有4p末端缺失及无WHS表型的个体来确定WHSCR的远端边界[South et al 2008a]。然而,由于发现了有更多远端缺失及核心表型(癫痫发作、生长发育迟缓或颅面特征)的WHS患者,目前的假设是, WHS是由接近基因组chr4: 419,224-2,010,962位置的1.6兆区域内基因缺失导致的邻近基因综合征。
WHSC1是一 新基因,横跨长90 kb的基因组区域,其中三分之二在WHSCR端粒末端。WHSC1在早期发育和蛋白质 结构域的选择性时空表达提示WHSC1可能在正常发育过程中发挥重要作用。它的 缺失可能导致WHS表型。然而,WHS表型严重程度的差异提示发挥作用的基因可能位于WHSCR的近端和远端,包括 NELFA (WHSC2)和LETM1[Zollino et al 2003, Bergemann et al 2005, Rodríguez et al 2005, South et al 2007].
NELFA(之前称WHSC2)参与 mRNA加工和细胞周期等多个方面。这个基因可能全面参与WHS[Kerzendorfer et al 2012].
LETM1是癫痫发作的候选基因,几乎所有患病个体都有LETM1缺失。LETM1蛋白在与细胞信号转导和能量生产有关的离子交换中发挥作用 [Nowikovsky et al 2004, Schlickum et al 2004, Hasegawa & van der Bliek 2007, Dimmer et al 2008, Jiang et al 2009, Kuum et al 2012]。然而,已观察到,包含 LETM1的4号染色体短臂(4P) 缺失个体并没有发生癫痫,且癫痫发作个体有一个不包含LETM1的4号染色体短臂(4P) 缺失[Van Buggenhout et al 2004, Faravelli et al 2007, Maas et al 2008, Misceo et al 2012, Bayindir et al 2013, Andersen et al 2014]。因此,很可能LETM1并不是导致癫痫发作的唯一基因。
仍需要大量的工作来明确正常生长发育个体及WHS个体中WHSC1和 LETM1的功能,以及描述WHSCR区域内基因和周边基因对临床表型的影响。此外,仅这两个基因的缺失不足以导致WHS表型 [Andersen et al 2014]。该区域内已明确与WHS表型相关的基因有:CTBP1、 CPLX1 和 PIGG与癫痫有关 [Misceo et al 2012, Bayindir et al 2013, Zollino et al 2014] , FGFRL1与颅面部特征及其他一些骨骼特征有关[Engbers et al 2009, Catela et al 2009, Hammond et al 2012]。
参考文献
Literature Cited
- Andersen EF, Carey JC, Earl DL, Corzo D, Suttie M, Hammond P, South ST. Deletions involving genes WHSC1 and LETM1 may be necessary, but are not sufficient to cause Wolf-Hirschhorn Syndrome. Eur J Hum Genet. 2014;22:464-70. [PMC free article: PMC3953918] [PubMed: 23963300]
- Antonius T, Draaisma J, Levichenko E, Knoers N, Renier W, van Ravenswaaij C. Growth charts for Wolf-Hirschhorn (4p-) syndrome (0-4 years of age). Eur J Pediatr. 2008;167:807-10. [PMC free article: PMC2413080] [PubMed: 17874131]
- Bailey NG, South ST, Hummel M, Wenger SL. Case report: cytogenetic and molecular analysis of proximal iInterstitial deletion of 4p, review of the literature and comparison with Wolf-Hirschhorn syndrome. J Assoc Genet Technol. 2010;36:5-10. [PubMed: 20177142]
- Battaglia A (2010) Deletion 4p - Wolf-Hirschhorn syndrome. In: Cassidy SB, Allanson JE, eds. Management of Genetic Syndromes. Hoboken, NJ: Wiley-Liss and Sons Inc. 249-61.
- Battaglia A, Carey JC. Update on the clinical features and natural history of Wolf Hirschhorn syndrome (WHS): experience with 48 cases. Am J Hum Genet. 2000;67:127.
- Battaglia A, Carey JC. Seizure and EEG patterns in Wolf-Hirschhorn (4p-) syndrome. Brain Dev. 2005;27:362-4. [PubMed: 16023553]
- Battaglia A, Carey JC, Cederholm P, Viskochil DH, Brothman AR, Galasso C. Natural history of Wolf-Hirschhorn syndrome: experience with 15 cases. Pediatrics. 1999a;103:830-6. [PubMed: 10103318]
- Battaglia A, Carey JC, Cederholm P, Viskochil DH, Brothman AR, Galasso C. Storia naturale della sindrome di Wolf-Hirschhorn: esperienza con 15 casi. Pediatrics. 1999b;11:236-42.
- Battaglia A, Carey JC, Viskochil DH, Cederholm P, Opitz JM. Wolf-Hirschhorn syndrome (WHS): a history in pictures. Clin Dysmorphol. 2000;9:25-30. [PubMed: 10649793]
- Battaglia A, Carey JC, Wright TJ. Wolf-Hirschhorn (4p-) syndrome. Adv Pediatr. 2001;48:75-113. [PubMed: 11480768]
- Battaglia A, Filippi T, Carey JC. Update on the clinical features and natural history of Wolf-Hirschhorn (4p-) syndrome: experience with 87 patients and recommendations for routine health supervision. Am J Med Genet Part C Semin Med Genet. 2008;148C:246-51. [PubMed: 18932224]
- Battaglia A, Filippi T, South ST, Carey JC. Spectrum of epilepsy and EEG patterns in Wolf-Hirschhorn syndrome: experience with 87 patients. Dev Med Child Neurol. 2009;51:373-80. [PubMed: 19379291]
- Battaglia A, South S, Carey JC. Clinical utility gene card for: Wolf-Hirschhorn (4p-) syndrome. Eur J Hum Genet. 2011 Apr;19(4) [PMC free article: PMC3060313] [PubMed: 21150881]
- Bayindir B, Piazza E, Della Mina E, Limongelli I, Brustia F, Ciccone R, Veggiotti P, Zuffardi O, Dehghani MR. Dravet phenotype in a subject with a der(4)t(4;8)(p16.3;p23.3) without the involvement of the LETM1 gene. Eur J Med Genet. 2013;56:551-5. [PubMed: 23999105]
- Bergemann AD, Cole F, Hirschhorn K. The etiology of Wolf-Hirschhorn syndrome. Trends Genet. 2005;21:188-95. [PubMed: 15734578]
- Calhoun ARUL, Carey JC, Kleimola C, Lortz A. Do patients with 4p- (Wolf-Hirschhorn syndrome) have an increased risk of liver carcinoma? Mont-Tremblant, Québec, Canada: 34th Annual David W. Smith Workshop on Malformations and Morphogenesis. 2013.
- Catela C, Bilbao-Cortes D, Slonimsky E, Kratsios P, Rosenthal N, te Welscher P. Multiple congenital malformations of Wolf-Hirschhorn syndrome are recapitulated in Fgfrl1 null mice. Dis Model Mech. 2009;2:283-94. [PMC free article: PMC2675798] [PubMed: 19383940]
- Chen CP, Hsu CY, Lee CC, Chen WL, Chen LF, Wang W. Prenatal diagnosis of de novo pure partial monosomy 4p (4p15.1-->pter) in a growth-restricted fetus with a Greek warrior helmet face and unilateral facial cleft on three-dimensional ultrasound. Prenat Diagn. 2004;24:934-6. [PubMed: 15565589]
- Curfs LM, Didden R, Sikkema SP, De Die-Smulders CE. Management of sleeping problems in Wolf-Hirschhorn syndrome: a case study. Genet Couns. 1999;10:345-50. [PubMed: 10631921]
- Dimmer KS, Navoni F, Casarin A, Trevisson E, Endele S, Winterpacht A, Salviati L, Scorrano L. LETM1, deleted in Wolf-Hirschhorn syndrome is required for normal mitochondrial morphology and cellular viability. Hum Mol Genet. 2008;17:201-14. [PubMed: 17925330]
- Engbers H, van der Smagt JJ, van ‘t Slot R, Vermeesch JR, Hochstenbach R, Poot M. Wolf-Hirschhorn syndrome facial dysmorphic features in a patient with a terminal 4p16.3 deletion telomeric to the WHSCR and WHSCR 2 regions. Eur J Hum Genet. 2009;17:129-32. [PMC free article: PMC2985965] [PubMed: 18830230]
- Faravelli F, Murdolo M, Marangi G, Bricarelli FD, Di Rocco M, Zollino M. Mother to son amplification of a small subtelomeric deletion: a new mechanism of familial recurrence in microdeletion syndromes. Am J Med Genet A. 2007;143A:1169-73. [PubMed: 17480006]
- Ferrarini A, Selicorni G, Cagnoli M, Zollino M, Lecce R, Chines C, Battaglia A. Distinct facial dysmorphism, pre and postnatal growth retardation, microcephaly, seizures. Mental retardation and hypotonia. Ital J Pediatr. 2003;29:393-7.
- Fisch GS, Grossfeld P, Falk R, Battaglia A, Youngblom J, Simensen R. Cognitive-behavioral features of Wolf-Hirschhorn syndrome and other subtelomeric microdeletions. Am J Med Genet. 2010;154C:417-26. [PubMed: 20981770]
- Grisaru S, Ramage IJ, Rosenblum ND. Vesicoureteric reflux associated with renal dysplasia in the Wolf-Hirschhorn syndrome. Pediatr Nephrol. 2000;14:146-8. [PubMed: 10684366]
- Hammond P, Hannes F, Suttie M, Devriendt K, Vermeesch JR, Faravelli F, Forzano F, Parekh S, Williams S, McMullan D, South ST, Carey JC, Quarrell O. Fine-grained facial phenotype-genotype analysis in Wolf-Hirschhorn syndrome. European journal of human genetics. EJHG. 2012;20:33-40. [PMC free article: PMC3234504] [PubMed: 21792232]
- Hanley-Lopez J, Estabrooks LL, Stiehm R. Antibody deficiency in Wolf-Hirschhorn syndrome. J Pediatr. 1998;133:141-3. [PubMed: 9672528]
- Hasegawa A, van der Bliek AM. Inverse correlation between expression of the Wolfs Hirschhorn candidate gene Letm1 and mitochondrial volume in C. elegans and in mammalian cells. Hum Mol Genet. 2007;16:2061-71. [PubMed: 17606466]
- Jiang D, Zhao L, Clapham DE. Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science. 2009;326:144-7. [PMC free article: PMC4067766] [PubMed: 19797662]
- Kagitani-Shimono K, Imai K, Otani K, Kamio N, Okinaga T, Toribe Y, Suzuki Y, Ozono K. Epilepsy in Wolf-Hirschhorn syndrome (4p-). Epilepsia. 2005;46:150-5. [PubMed: 15660782]
- Kerzendorfer C, Hannes F, Colnaghi R, Abramowicz I, Carpenter G, Vermeesch JR, O'Driscoll M. Characterizing the functional consequences of haploinsufficiency of NELF-A (WHSC2) and SLBP identifies novel cellular phenotypes in Wolf-Hirschhorn syndrome. Hum Mol Genet. 2012;21:2181-93. [PubMed: 22328085]
- Kuum M, Veksler V, Liiv J, Ventura-Clapier R, Kaasik A. Endoplasmic reticulum potassium-hydrogen exchanger and small conductance calcium-activated potassium channel activities are essential for ER calcium uptake in neurons and cardiomyocytes. Journal of cell science. 2012;125:625-33. [PubMed: 22331352]
- Maas NM, Van Buggenhout G, Hannes F, Thienpont B, Sanlaville D, Kok K, Midro A, Andrieux J, Anderlid BM, Schoumans J, Hordijk R, Devriendt K, Fryns JP, Vermeesch JR. Genotype-phenotype correlation in 21 patients with Wolf-Hirschhorn syndrome using high resolution array comparative genome hybridisation (CGH). J Med Genet. 2008;45:71-80. [PubMed: 17873117]
- Meloni A, Shepard RR, Battaglia A, Wright TJ, Carey JC. Wolf-Hirschhorn syndrome: correlation between cytogenetics, FISH, and severity of disease. Am J Hum Genet. 2000;67:149.
- Misceo D, Barøy T, Helle JR, Braaten O, Fannemel M, Frengen E. 1.5Mb deletion of chromosome 4p16.3 associated with postnatal growth delay, psychomotor impairment, epilepsy, impulsive behavior and asynchronous skeletal development. Gene. 2012;507:85-91. [PubMed: 22842548]
- Nowikovsky K, Froschauer EM, Zsurka G, Samaj J, Reipert S, Kolisek M, Wiesenberger G, Schweyen RJ. The LETM1/YOL027 gene family encodes a factor of the mitochondrial K+ homeostasis with a potential role in the Wolf-Hirschhorn syndrome. J Biol Chem. 2004;279:30307-15. [PubMed: 15138253]
- Prunotto G, Cianci P, Cereda A, Scatigno A, Fossati C, Maitz S, Biondi A, Selicorni A. Two cases of hepatic adenomas in patients with Wolf-Hirschhorn syndrome: a new rare complication? Am J Med Genet A. 2013;161A:1759-62. [PubMed: 23696331]
- Rodríguez L, Zollino M, Climent S, Mansilla E, López-Grondona F, Martínez-Fernández ML, Murdolo M, Martínez-Frías ML. The new Wolf-Hirschhorn syndrome critical region (WHSCR-2): a description of a second case. Am J Med Genet A. 2005;136:175-8. [PubMed: 15948183]
- Schlickum S, Moghekar A, Simpson JC, Steglich C, O'Brien RJ, Winterpacht A, Endele SU. LETM1, a gene deleted in Wolf-Hirschhorn syndrome, encodes an evolutionarily conserved mitochondrial protein. Genomics. 2004;83:254-61. [PubMed: 14706454]
- Shanske AL, Yachelevich N, Ala-Kokko L, Leonard J, Levy B. Wolf-Hirschhorn syndrome and ectrodactyly: new findings and a review of the literature. Am J Med Genet Part A. 2010;152A:203-8. [PubMed: 20034099]
- Sharathkumar A, Kirby M, Freedman M, Abdelhaleem M, Chitayat D, Teshima IE, Dror Y. Malignant hematological disorders in children with Wolf-Hirschhorn syndrome. Am J Med Genet A. 2003;119A:194-9. [PubMed: 12749063]
- South ST, Bleyl SB, Carey JC. Two unique patients with novel microdeletions in 4p16.3 that exclude the WHS critical regions: implications for critical region designation. Am J Med Genet A. 2007;143A:2137-42. [PubMed: 17696124]
- South ST, Hannes F, Fisch GS, Vermeesch JR, Zollino M. Pathogenic significance of deletions distal to the currently described Wolf-Hirschhorn syndrome critical regions on 4p16.3. Am J Med Genet C Semin Med Genet. 2008a;148C:270-4. [PubMed: 18932125]
- South ST, Rope AF, Lamb AN, Aston E, Glaus N, Whitby H, Maxwell T, Zhu XL, Brothman AR. Expansion in size of a terminal deletion: a paradigm shift for parental follow-up studies. J Med Genet. 2008b;45:391-5. [PubMed: 18413369]
- South ST, Whitby H, Battaglia A, Carey JC, Brothman AR. Comprehensive analysis of Wolf-Hirschhorn syndrome using array CGH indicates a high prevalence of translocations. Eur J Hum Genet. 2008c;16:45-52. [PubMed: 17726485]
- Ulualp SO, Wright CG, Pawlowski KS, Roland PS. Histopathological basis of hearing impairment in Wolf-Hirschhorn syndrome. Laryngoscope. 2004;114:1426-30. [PubMed: 15280721]
- Van Buggenhout G, Melotte C, Dutta B, Froyen G, Van Hummelen P, Marynen P, Matthijs G, de Ravel T, Devriendt K, Fryns JP, Vermeesch JR. Mild Wolf-Hirschhorn syndrome: micro-array CGH analysis of atypical 4p16.3 deletions enables refinement of the genotype-phenotype map. J Med Genet. 2004;41:691-8. [PMC free article: PMC1735886] [PubMed: 15342700]
- Wieczorek D, Krause M, Majewski F, Albrecht B, Horn D, Riess O, Gillessen-Kaesbach G. Effect of the size of the deletion and clinical manifestation in Wolf-Hirschhorn syndrome: analysis of 13 patients with a de novo deletion. Eur J Hum Genet. 2000;8:519-26. [PubMed: 10909852]
- Wright TJ, Ricke DO, Denison K, Abmayr S, Cotter PD, Hirschhorn K, Keinanen M, McDonald-McGinn D, Somer M, Spinner N, Yang-Feng T, Zackai E, Altherr MR. A transcript map of the newly defined 165 kb Wolf-Hirschhorn syndrome critical region. Hum Mol Genet. 1997;6:317-24. [PubMed: 9063753]
- Wu-Chen WY, Christiansen SP, Berry SA, Engel WK, Fray KJ, Summers CG. Ophthalmic manifestations of Wolf-Hirschhorn syndrome. J AAPOS. 2004;8:345-8. [PubMed: 15314595]
- Zollino M, Di Stefano C, Zampino G, Mastroiacovo P, Wright TJ, Sorge G, Selicorni A, Tenconi R, Zappala A, Battaglia A, Di Rocco M, Palka G, Pallotta R, Altherr MR, Neri G. Genotype-phenotype correlations and clinical diagnostic criteria in Wolf-Hirschhorn syndrome. Am J Med Genet. 2000;94:254-61. [PubMed: 10995514]
- Zollino M, Lecce R, Fischetto R, Murdolo M, Faravelli F, Selicorni A, Butte C, Memo L, Capovilla G, Neri G. Mapping the Wolf-Hirschhorn syndrome phenotype outside the currently accepted WHS critical region and defining a new critical region, WHSCR-2. Am J Hum Genet. 2003;72:590-7. [PMC free article: PMC1180235] [PubMed: 12563561]
- Zollino M, Lecce R, Selicorni A, Murdolo M, Mancuso I, Marangi G, Zampino G, Garavelli L, Ferrarini A, Rocchi M, Opitz JM, Neri G. A double cryptic chromosome imbalance is an important factor to explain phenotypic variability in Wolf-Hirschhorn syndrome. Eur J Hum Genet. 2004;12:797-804. [PubMed: 15241479]
- Zollino M, Murdolo M, Marangi G, Pecile V, Galasso C, Mazzanti L, Neri G. On the nosology and pathogenesis of Wolf-Hirschhorn syndrome: genotype-phenotype correlation analysis of 80 patients and literature review. Am J Med Genet C Semin Med Genet. 2008;148C:257-69. [PubMed: 18932124]
- Zollino M, Orteschi D, Ruiter M, Pfundt R, Steindl K, Cafiero C, Ricciardi S, Contaldo I, Chieffo D, Ranalli D, Acquafondata C, Murdolo M, Marangi G, Asaro A, Battaglia D. Unusual 4p16.3 deletions suggest an additional chromosome region for the Wolf-Hirschhorn syndrome-associated seizures disorder. Epilepsia. 2014;55:849-57. [PubMed: 24738919]
Chapter Notes
Author History
Agatino Battaglia, MD (2001-present)
John C Carey, MD (2001-present)
Sarah T South, PhD (2001-present)
Tracy J Wright, PhD; Hunter Animal Hospital (2001-2015)
Revision History
- 20 August 2015 (me) Comprehensive update posted live
- 17 June 2010 (me) Comprehensive update posted live
- 24 March 2009 (cd) Revision: deletion/duplication analysis available clinically
- 25 September 2006 (me) Comprehensive update posted to live Web site
- 6 April 2004 (me) Comprehensive update posted to live Web site
- 29 April 2002 (me) Review posted to live Web site
- 2 February 2001 (ab) Original submission