
简介
临床特征 混合性卟啉病(VP)既是皮肤卟啉病(具有慢性水疱性皮肤病变)又是急性卟啉病(具有严重的发作性神经精神症状)。 VP的最常见表现是成年期暴露于阳光下的皮肤(尤其是手和脸)皮肤起水疱(表皮下的囊泡,大疱和侵蚀逐渐形成至愈合)。其他慢性皮肤病表现包括丘疹 ,疤痕,增厚以及皮肤色素沉着减少和增加,可能会出现面部色素沉着和过度发汗。皮肤的表现在冬季可能有所改善,在北部地区和深色皮肤的人中较少见。青春期后的任何时间都可能出现急性神经精神症状,但老年人中的发病率却较低。急性表现变化很大,但对于反复发作的人,每次发作可能相似。并非所有的表现都出现在每次发作中;急性症状可能会变为慢性。女性比男性更容易出现症状。最常见的表现是腹痛;便秘;背部、胸部和四肢疼痛;焦虑;癫痫发作并且主要是运动神经病,导致肌肉无力,可能会发展为四肢瘫痪和呼吸麻痹。还可以观察到精神障碍和自主神经病变。急性发作可能很严重,甚至可能致命。
诊断/测试 VP的生化诊断是在尿液中胆卟啉原(PBG)或卟啉升高且血浆荧光扫描的〜626 nm处出现荧光峰的患者中建立的;粪卟啉也升高,主要是粪卟啉III和原卟啉。 VP的分子诊断是通过在分子遗传学检测上鉴定PPOX中的 杂合的 致病性变异而建立的。
管理 表型的治疗:治疗急性神经内脏发作或皮肤表现的第一步是识别并清除恶化的因素(请参阅:避免的药物/情况)。大多数急性神经内脏发作需要入院;癫痫发作,运动神经病和低钠血症的存在提示严重疾病,理想情况下应在ICU中进行管理。通常需要止痛药来止痛。恩丹西酮或相关药物可用于恶心和呕吐;吩噻嗪可有效治疗恶心,躁动和幻觉。
尽管有时可以在门诊患者中使用葡萄糖负荷治疗轻度发作(无癫痫发作,无力或低钠血症且不需要麻醉药),但大多数发作都需要静脉血红素治疗和住院观察以进一步支持治疗。
皮肤表现最好通过穿防护服并避免暴露在阳光下来预防。消除诱发因素后,症状可能会减轻。尚无有效降低卟啉水平和减轻皮肤症状的治疗方法。对于疼痛的病灶可能需要使用止痛药,对于合并感染可能需要使用抗生素。
预防主要表现:如果纠正或避免诱发因素,则不太可能发生急性神经内脏症状。促性腺激素释放激素类似物可预防复发的急性发作。每周或每两周一次的血红素输注以预防频繁的非周期性发作可能是有效的,但缺乏经验。预防皮肤表现需要防晒。
监测:从50岁开始,在胆色素原或卟啉持续升高的人群中,每六个月进行一次肝脏成像,可以发现早期肝细胞癌。
避免的药物/情况:应避免的加剧因素包括以下药物:巴比妥类药物,磺胺类抗生素,灰黄霉素,利福平,大多数抗惊厥药,包括苯妥英和卡马西平,酒精,麦角麦角生物碱,胃复安和孕激素。尽管通常应避免使用避孕药,但可以耐受低剂量的激素制剂。伴随疾病应使用尽可能被认为安全的药物进行有效治疗。美国卟啉基金会和欧洲卟啉网的网站上保存了更新的安全和不安全药物清单。
对有风险的亲属的评估:可以为有风险的家庭成员提供针对特定家庭的PPOX致病性变异的分子遗传学检测 ,以识别那些杂合的个体(目的是为了合理使用药物和避免恶化因素)。生化测试,尤其是血浆荧光扫描和粪便卟啉分析也很有用,但它不如分子遗传测试灵敏。
妊娠管理:精氨酸血红素或氢氧化血红素(血红素)已成功治疗了妊娠期发作。尽管两种制剂都没有在怀孕期间进行过广泛的研究,但多年的经验表明,怀孕期间的治疗不太可能对胎儿产生不良影响。
遗传咨询 VP以 常染色体显性遗传方式遗传, 外显率降低。新发致病性变异很少见。具有VP的个人的每个孩子都有50%的风险遗传致病性变异;继承了该变异的后代可能会或可能不会出现表现,但大多数不会。如果已经确定了受累的家属的致病变异,则可能对高VP风险进行产前检查。值得注意的是,PPOX致病变异的存在不能预测一个人是否会出现症状(以及在多大年龄出现)。
诊断
提示性发现
具有以下临床发现和初步实验室发现的个体应怀疑混合性卟啉病(VP)。
临床发现
- 皮肤表现包括慢性起泡,光敏性,最常见于手背。慢性特征包括水疱,粟粒,疤痕,增厚以及皮肤色素沉着减少和增加。可能会出现面部色素沉着和过度发汗。皮肤病变与皮肤卟啉病(porphyria cutanea tarda,PCT)和其他起泡的皮肤卟啉病相同[Meissner et al 2003](请参阅Differential Diagnosis)。
- 神经内脏症状最常见的有以下几种:
- 腹痛。疼痛通常是严重的,钝痛而不是绞痛,并且广泛而不是局部的。由于疼痛是神经性而非炎性的,因此与疼痛的严重程度相比,腹部的发现微乎其微。可能出现肠梗阻和膀胱膨胀。初诊后因常见原因仍无法解释腹痛时,应怀疑是急性肝卟啉病。
- 便秘
- 背部,胸部和四肢疼痛
- 焦虑
- 癫痫发作
- 由于主要是运动神经病引起的肌肉无力,通常始于近端上肢,可能发展为四肢瘫痪和呼吸麻痹。这伴随着疼痛,有时甚至是感觉丧失。最初可能会出现反射亢进,然后随着运动神经病的发展而出现反射减退。
- 低钠血症,增加癫痫发作的风险。它可能是下丘脑受累的表现和抗利尿激素分泌不当的综合征 [Anderson et al 2005].
最初的生化实验室发现。由于VP可能会在阳光暴晒的皮肤上出现水疱性皮肤损伤,神经内脏症状或同时出现这两种情况,因此最初的一线测试旨在检测可能导致皮肤或神经内脏表现的所有卟啉病(请参见 Differential Diagnosis)。
- 皮肤起泡卟啉症(包括VP)。当怀疑是VP或任何其他起泡的皮肤卟啉症时,建议的初始测试是测量血浆或尿卟啉。如果升高,则需要进一步测试以确定卟啉的类型,或者卟啉升高(特别是在尿液中)是否代表非特异性卟啉尿 。
- 急性卟啉病(包括VP)。测量尿中的胆色素原(PBG)*和总卟啉。尿中的δ-氨基乙酰丙酸(ALA)通常与PBG同时测定,但这对于初次筛查不是必需的。*注:(1)如果尿中PBG明显升高证实为急性卟啉病,可以在适当的情况下开始治疗急性发作的症状(请参阅管理,Treatment of Manifestations),同时进行进一步的生化测试以确定急性卟啉病的类型(请参阅Differential Diagnosis)。 (2)如果PBG正常,则应在同一尿液样本中测量总卟啉和ALA,因为总卟啉的升高时间通常比PBG长。在ALA脱水酶缺乏症的卟啉症(ADP)中,稀有的卟啉,ALA和总卟啉(而非PBG)显著升高[Anderson et al 2005].
红细胞卟啉的实质性升高与VP不一致,并提示红细胞性卟啉病是皮肤出现水疱,尿液和血浆卟啉升高的原因。相应地,患有VP的个体中大量的红细胞原卟啉可能提示同时存在高原卟啉锌的状况,例如铁缺乏症,铅中毒或另一种红细胞疾病。
建立诊断
生化诊断 当最初的生化实验室发现支持急性卟啉病(即尿中PBG或卟啉升高)或皮肤起泡性卟啉病(即血浆或尿中卟啉升高)时,需要进一步的诊断性生化试验(Table 1)以将VP与其他急性和慢性皮肤卟啉症和导致非特异性卟啉尿的疾病鉴别(例如肝脏疾病):
- 血浆荧光扫描 当尿中PBG升高时,血浆荧光扫描可以建立或排除VP,因为在任何其他类型的卟啉症中均未发现〜626 nm的荧光峰。
- 粪卟啉分析 可以区分VP,acute intermittent porphyria(急性间歇性卟啉症,AIP)和hereditary coproporphyria(遗传性卟啉症,HCP),这是使尿PBG显著升高的唯一疾病。
- 粪卟啉分析和血浆荧光扫描 还可以可靠地将VP与porphyria cutanea tarda(迟发性皮肤卟啉病 ,PCT)和其他引起皮肤损伤的卟啉区分开(请参阅Differential Diagnosis)。
Table 1
混合性卟啉病(VP)的生化特性
| 缺陷酶 | 尿PBG和卟啉 | 血浆荧光扫描 | 粪卟啉 | |||
|---|---|---|---|---|---|---|
| Active | Asx | Active | Asx | Active | Asx | |
| PPOX 1, 2 | ↑ PBG, ALA & 总卟啉 3, 4, 5 | ↑ or NI PBG, ALA &总卟啉 6 | ↑; 见注脚 8 | ↑; 见注脚 8 | 见注脚 7 | 见注脚 8 |
Active=有症状的PPOX杂合子; ALA =δ-氨基乙酰丙酸; Asx =无症状的PPOX杂合子; NI =不增加; PBG =胆色素原PPOX =原卟啉原氧化酶
- 1.
该酶将原卟啉原氧化为原卟啉,其缺乏导致原卟啉原在肝脏中的积累,随后其被自氧化为原卟啉。
- 2.
酶测定不需要用于诊断目的,并且不能广泛使用。
- 3.
PBG升高应通过定量方法检测,例如 Mauzerall & Granick [1956]描述的方法,该方法也可测量ALA或质谱。定性方法(如Watson-Schwartz和Hoesch检验)的结果被认为已过时,应通过定量方法对同一样品进行确认。 ALA的水平低于PBG。注意:ALAD卟啉症(ADP)中的ALA升高,其中PBG正常或仅轻微升高。
- 4.
Active VP是由定量PBG显著提高而确诊。
- 5.
对于筛查,在相同尿液样品中测量总卟啉也很有用,因为VP和HCP中的PBG水平可以比AIP中的PBG升高较少,并且可以更快地降低至正常水平。注意:与PBG的大幅增加不同,尿卟啉的大幅增加并不表示卟啉症,因为在许多其他医学状况下,尤其是肝胆系统或骨髓 受累的时,尿卟啉会增加。
- 6.
症状缓解的人的PBG和总卟啉可能不会升高。如果怀疑急性卟啉症已引起过去的症状,则可能需要进行全面的生化检查,包括尿液ALA,PBG和卟啉,粪便卟啉和血浆卟啉。
- 7.
粪卟啉在HCP和VP中显著升高,而在AIP中则很少或没有升高。粪卟啉的类型区分HCP和VP,在HCP中伴有卟啉III的明显优势,而在VP中卟啉III和原卟啉的水平升高了。
- 8.
稀释血浆的荧光扫描在中性pH值下可在VP中〜626 nm波长处获得一个荧光峰,该峰对该卟啉具有高度敏感性和特异性[Poh-Fitzpatrick 1980]。这是在没有症状的情况下建立VP的最灵敏的生化方法。粪卟啉分析的灵敏度低于血浆荧光扫描。
分子诊断 致病性变异的鉴定现在被认为是VP和其他急性卟啉症的治疗标准,以确认诊断并告知遗传咨询(请参阅Genetic Counseling)(请参阅选项1和选项2)。
选项1.通常最好先建立VP的生化诊断,然后进行确证的单基因 (PPOX)检测。但是,当生化检测(例如,PBG显著升高)表明诊断为AIP,HCP或VP或待检测的个体无症状时,没有或没有特异性的生化异常,可以使用multigene panel(多基因套餐, HMBS,CPOX,PPOX)建立诊断。
- 通常建议在生化诊断VP后对PPOX进行单基因检测。序列分析可检测出较小的基因内缺失/插入以及错义, nonsense和 剪接位点变异;通常,不会检测到外显子或全基因缺失/重复。标准做法是先执行 序列分析。如果在经生化证明具有VP的个体中未发现PPOX致病性变异,请进行基因靶向的 deletion/duplication analysis 以检测基因内的缺失或重复。
注意:当在南非的Afrikaner人群中以生物化学方法建立VP时,可以考虑针对该人群中约95%的混合卟啉病患者 建立者变异, p.Arg59Trp
进行针对性分析 [Dean 1971, Meissner et al 1996]。参见Table 4. Notable PPOX Pathogenic Variants。
- 包含PPOX和其他感兴趣的基因(尤其是HMBS和CPOX;请参见“鉴别诊断”)的急性卟啉病multigene panel 最有可能以最合理的成本确定症状和PBG升高的遗传原因,同时减少了意义不确定性变异和不能解释潜在表型的基因中的致病性变异的出现。
ALAD,编码ALA脱水酶(缺乏ALA脱水酶缺陷型卟啉症)的基因也可包括在套餐中,但仅在ALA和卟啉(但不包括PBG)升高时才相关。
注意:(1)每个基因所用测试的诊断敏感性可能因实验室而异,并可能随时间而变化。 (2)套餐中使用的方法可能包括序列分析, deletion/duplication analysis和/或其他非基于序列的测试。
有关多基因套餐的介绍,请单击here。有关订购基因检测的临床医生的更多详细信息,请参见here。
选项2 不建议将基因组测试用于急性卟啉病的初步诊断。但是,当无法解释症状原因,鉴别HMBS,CPOX或PPOX的致病性变异或意义不确定性的变异时,建议基因组的检测。PBG和卟啉升高的生化检验证实了卟啉症–并确认VP的诊断。
有关全面的 基因组的测试的介绍,请单击here。可在here找到有关订购基因组测试的临床医生的更多详细信息。
Table 2
混合性急性卟啉病的分子遗传学检测
| 基因 1 | 方法 | 检测方法可的具有致病变异2 先证者所占比例 |
|---|---|---|
| PPOX | 测序 3 | 96%-100% 4 |
| 基因靶向 deletion/duplication analysis 5 | Unknown 6 | |
| 致病性变异的靶向分析 | p.Arg59Trp 7 |
- 1.
染色体 位点 和蛋白见 Table A. Genes and Databases .
- 2.
有关等位基因变异的信息见 Molecular Genetics.
- 3.
- 4.
- 5.
目标基因deletion/duplication analysis 鉴定基因内缺失或扩增。可采用的方法有 : quantitative PCR, 长链PCR, 多重连接依赖性探针扩增 (MLPA), 目的 基因 微阵列可检出单个 外显子缺失和扩增。
- 6.
已经报道了PPOX的多外显子缺失[Barbaro et al 2013]。 但是,目前尚无有关基因靶向的deletion/duplication analysis检测率的数据。
- 7.
VP在南非尤为普遍, 建立者变异 p.Arg59Trp [Dean 1971]约占病例的95% [Meissner et al 1996]。
临床特征
临床表现
混合性卟啉病(VP)分为皮肤卟啉病和急性卟啉病。它可以表现为慢性水疱性皮肤表现和/或可能演变为慢性的神经内脏表现的急性发作。
皮肤表现 VP的最常见表现是慢性起泡,光敏性,通常出现在手背。损伤是由阳光照射引起的,该阳光激活卟啉并使皮肤脆弱并易于形成水疱。病变位于受阳光照射的区域,尤其是手背,而较少出现在面部,颈部,耳朵和下肢。由于阳光引起的损害不是很严重,因此通常认识不到阳光的作用。皮肤的表现在冬季可能有所改善,在北部地区和深色皮肤的人中较不普遍。
VP的这些和其他表现通常出现在成年期,很少出现在青春期之前。
表皮下的囊泡,大疱和糜烂结痂并缓慢愈合。当水泡破裂时,它们可能会感染并引起痛苦。
其他慢性皮肤病表现包括粟粒,疤痕,增厚以及皮肤色素沉着减少和增加的区域。可能会出现面部色素沉着和过度发汗。
皮肤表现与迟发性皮肤卟啉病( porphyria cutanea tarda,PCT)和遗传性卟啉病 hereditary coproporphyria,HCP)相同,但不如先天性红细胞生成性卟啉症( congenital erythropoietic porphyria,CEP)和肝造血性卟啉症(hepatoerythropoietic porphyria,HEP)明显。它们与红细胞生成性原卟啉症(erythropoietic protoporphyria,EPP)的急性非起泡性光皮肤表现形成对照(见Table 3)。
值得一提的是,绝大多数杂合的PPOX致病性变异的人都是无症状的,除非VP家族史进行筛查,否则不太可能被识别(请参阅Genetic Counseling)。在南非,近几十年来,急性发作的频率有所下降。这可能是由于在临床实践中较少使用诸如巴比妥类药物和磺酰胺类抗生素之类的有害药物,以及可能更好地识别病例和更好地宣传有关如何避免未来发作的信息。 VP现在在南非更常见,表现为皮肤而非急性表现[Meissner et al 2003, Anderson et al 2005, Hift & Meissner 2005].
神经内脏症状可在青春期后的任何年龄发生,都可能是急性发作,但可能会变得慢性。女性的症状比男性更常见,而老年人的症状则更少。发作的频率和严重程度相差很大,并且在一定程度上取决于诱发因素,例如某些药物,激素和营养不足[Anderson et al 2005]。经历急性发作的杂合的PPOX 致病性变异的人的比例从1980年代的30%-40%下降到2005年的5%-10% [Hift & Meissner 2005].
神经内脏症状与其他急性卟啉症相同(请参见Differential Diagnosis)。
急性表现各不相同。最常见的症状是腹痛,恶心和呕吐;便秘;背部,胸部和四肢疼痛;焦虑;癫痫发作,主要是运动性周围神经病变,导致肌肉无力,可能会发展为四肢瘫痪和呼吸麻痹[Kauppinen & Mustajoki 1992, Meissner et al 2003, Anderson et al 2005, Hift & Meissner 2005]。还可以观察到精神障碍和自主神经病变。并非所有症状都出现在单个发作中,并且症状在发作之间可能有所不同。但是,反复发作通常是相似的。急性发作可能很严重,甚至可能致命。但与急性间歇性卟啉症( acute intermittent porphyria ,AIP)相比,急性发作的频率较低,强度较低 [Hift & Meissner 2005].
运动神经病通常最初表现为近端上肢肌肉无力,可能难以发现。最初可能会出现反射亢进,然后随着运动神经病的发展而出现反射亢进。运动神经病可能伴有感觉丧失。注意:由于急性卟啉症引起的运动神经病伴有很少或没有脑脊液蛋白升高,这有助于将其与LandryGuillain-Barré综合征区分开 [Anderson et al 2005].
由于腹部疼痛是神经性而非炎性的,因此与疼痛的严重程度相比,腹部的发现微乎其微。可能出现肠梗阻和膀胱膨胀。
如果存在包括神经病,癫痫发作和呼吸系统损害在内的严重表现,急性发作可能是致命的。如果处理得当,急性发作的结果通常会很好。即使是剧烈的运动神经病,也可以在不同的月份(有时甚至是几年)内恢复,并且可以逆转。
经常被确定为易患急性发作的因素包括接触有害药物,饮酒,饮食摄入减少或感染或其他疾病引起的应急。已知大多数有害药物都是肝δ-氨基乙酰丙酸合酶(ALAS)和肝细胞色素P450酶的诱导剂(请参阅要Agents/Circumstances to Avoid)。怀孕通常可以很好地耐受,但在某些女性中会引发急性发作。
自主神经病和循环儿茶酚胺水平升高会导致诸如心动过速,高血压,躁动不安和躁动等体征。
慢性疼痛可能是VP和其他急性卟啉症的表现。抑郁症可能更难与疾病联系起来。慢性疼痛和抑郁可能成为重要的管理问题。
慢性肝异常,特别是血清转氨酶轻度升高是常见的。 VP(以及 AIP和 HCP)增加了发展为肝细胞癌和慢性肾脏疾病的风险。肝胆管癌可能会发展成肝癌,尤其是在50岁以后,胆色素原和卟啉持续升高的人。
注意:关于乔治三世国王(也许还有英国王室中的其他人)患 VP的猜测被轻视了 [Peters 2011].
基因型-表型相关
PPOX致病变异通常很严重,导致酶活性很少低,或者没有酶活性。剩余的大约一半的正常酶活性是正常等位基因的产物。因此,不同的致病变异与疾病严重程度的差异无关[Whatley et al 1999, Whatley et al 2009].
血红素生物合成途径中两个不同基因的致病变异的双重杂合性。在发现其他家庭成员具有VP的临床和生化特征后,最初被诊断为HCP的皮肤表现患者被发现是PPOX和CPOX致病变异的双重杂合子 [van Tuyll van Serooskerken et al 2011]. 。这种稀有的双杂合子的表型不一定比与仅一个 基因的致病性变异的杂合性相关的 表型严重,这表明双重致病性杂合的变异的卟啉症个体可能比预估的更为普遍。
注意:通常会因为不寻常的生化模式而怀疑具有双重杂合性,因此,如果不进行全面的生化测试,就不太可能被发现[van Tuyll van Serooskerken et al 2011],这表明需要额外的分子遗传学检测.
外显率
导致VP的PPOX致病变异几乎不产生功能性酶;正常残留酶活性的大约50%主要来自正常的等位基因。外显率低,但是可能会因增加对肝血红素合成的需求而增加。外显率可能受到修饰基因的影响,这些基因有待确定。
命名法
混合性卟啉症(VP)和遗传性卟啉症(hereditary coproporphyria,HCP)有时被称为混合卟啉症,现在已经过时了。
VP也被称为南非急性卟啉症或原卟啉症。
过去,在某些情况下,可能没有明显区分家族性/迟发性皮肤卟啉病( porphyria cutanea tarda,PCT)和VP。
患病率
据估计,在南非人口中,每千人中有3个人是PPOX 杂合的致病性变异 p.Arg59Trp [Meissner et al 1996, Meissner et al 2003].
在欧洲,有现在或过去症状的VP患病率约为急性间歇性卟啉症(AIP)的一半,估计为3.2:1,000,000 [Elder et al 2013].
遗传相关(等位基因)疾病
具有双等位基因的PPOX致病变异 (即纯合子或复合杂合子)的个体很少见[Frank et al 1998, Pinder et al 2013]。 在儿童时期,他们表现神经系统症状,包括智力残疾和/或癫痫发作以及仅皮肤表现。 在这些个体中,一个或两个等位基因必须产生某种PPOX酶。
鉴别诊断
遗传性卟啉病包括一组不同的疾病,每种疾病均是由于血红素合成途径中特定步骤的改变而导致的,该改变导致途径中间体的积累 (Figure 1).
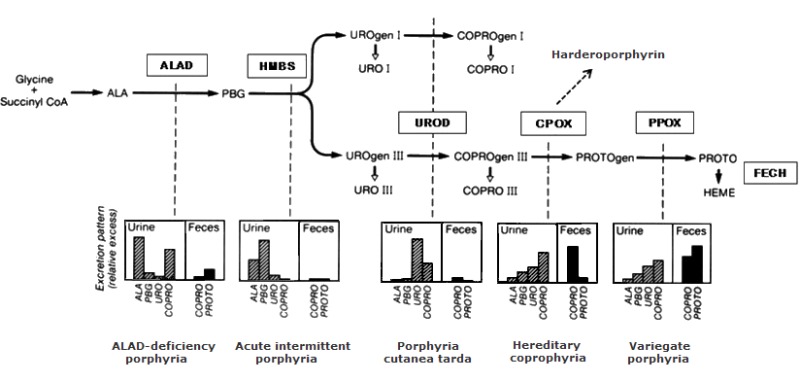
在 Table 3中,卟啉病按其主要临床表现(神经内脏或皮肤)和途径中间产物过量产生的组织来源进行分组:肝(即肝性); 或骨髓(即红细胞性)。
具有神经系统表现的卟啉病被认为是急性的,因为这些症状通常表现为多样的,严重的发作,这可能是由内源激素,药物和饮食变化引起的。由于它们的稀有性和症状的非特异性,即使严重也很难诊断。四种急性卟啉病(通常称为急性肝卟啉病)是:ALA脱水酶缺乏症卟啉病(ADP),急性间歇性卟啉病(acute intermittent porphyria,AIP),遗传性卟啉病(hereditary coproporphyria,HCP)和混合卟啉病(VP)。世界文献中仅报道了少数患有ADP的个体,尚不确定该卟啉症是肝性的,还是红细胞性或者两者兼有。
具有皮肤表现的卟啉病包括那些引起慢性水疱性皮肤病变的患者(即VP以及porphyria cutanea tarda (迟发性皮肤卟啉病 ,PCT),HCP,先天性红细胞生成性卟啉病(congenital erythropoietic porphyria,CEP)和肝性卟啉病[(hepatoerythropoietic porphyria,HEP)或急性非水疱性光敏(即EPP和XLP)。
Table 3
遗传性卟啉病的分类
| 卟啉病分类 | 基因(s) | MOI | 发现 | ||
|---|---|---|---|---|---|
| 神经内脏表现1 | 光敏性 | ||||
| 肝性 | ADP | ALAD | AR | + | 0 |
| AIP | HMBS | AD | + | 0 | |
| HCP | CPOX | AD | + | + | |
| PCT type II 2 | UROD | AD | 0 | + | |
| HEP 3 | AR | 0 | + | ||
| VP | PPOX | AD | + | + | |
| 红细胞性 | CEP 4 | UROS | AR | 0 | + |
| GATA1 | XL | ||||
| EPP | FECH | AR | 0 | + 5 | |
| XLP | ALAS2 | XL | 0 | + 5 | |
- 1.
具有神经内脏表现的卟啉病被认为是“急性的”,因为症状通常以多样,严重的发作急性发作。但是,一些受累的人会出现慢性表现。
- 2.
- 3.
- 4.
- 5.
与其他皮肤卟啉病(包括VP)的慢性起泡相反,EPP和XLP的光皮肤表现是急性且非起泡。
急性神经性卟啉病 VP的急性神经内脏症状与其他急性卟啉症相同。 VP可通过血浆荧光扫描和粪便卟啉分析 (Table 1) 或分子遗传学检测 (Table 2)与AIP和HCP区分。
在由于急性卟啉症之一(AIP,VP,HCP和ADP)引起的运动神经病而导致进行性无力的个体中,最可能被考虑的疾病是急性多发性神经病,即LandryGuillain-Barré综合征。
- 腹痛,便秘和心动过速通常伴有急性卟啉症的急性神经系统疾病,但LandryGuillain-Barré综合征不伴有。
- CSF蛋白通常在急性卟啉症中是正常的,但在LandryGuillain-Barré综合征中通常会升高。
- 最重要的是,在急性卟啉症中尿PBG明显升高,尤其是在出现症状时,但在LandryGuillain-Barré综合征中正常。
慢性水疱性皮肤卟啉病。通过生化测试可以很容易地将VP与以下区分开,并最终由分子遗传学检测确认。
- 最常见的人类卟啉病:迟发性皮肤卟啉病(porphyria cutanea tarda ,PCT)起泡的皮肤损伤与VP相同。因为PCT比VP更常见,所以VP患者常被误诊为PCT。由于PCT的治疗措施对VP无效,因此在开始治疗之前区分这些疾病非常重要。
- HCP与此类皮肤表现的关联要比VP少得多。
- 当并发的终末期肾脏疾病伴发卟啉排泄障碍而血浆卟啉水平升高时,AIP才会出现皮肤起泡现象。
- CEP和HEP的皮肤表现也慢性且起泡,但通常比VP更严重,因为循环中的卟啉水平通常比PCT和VP高得多(通常高一个数量级)。尽管在临床评估期间很容易将轻度CEP和HEP患者的诊断误认为VP,HCP和PCT,但这些红细胞生成性卟啉症尤其是通过发现高水平的红细胞卟啉来区分。
- 假性卟啉病是一种尚不清楚的疾病,其皮肤表现类似于PCT和VP,但卟啉没有明显升高。
处理
初步诊断后的评估
为了确定疾病的程度并规划混合性卟啉症(VP)的个体的治疗,建议进行以下临床和实验室评估(如果未作为导致诊断的评估的一部分进行):
- 确定血浆和尿卟啉和尿卟啉胆原原(PBG)的升高程度,如果在诊断时未测定
- 对当前任何急性神经内脏表现进行临床评估,以确定是否需要住院和使用血红素治疗
- 神经系统. 评估引起轻瘫,疼痛或感觉改变的神经系统受累程度
- 精神病学评估. 如果存在抑郁或其他精神病学特征
- 肝. 肝功能检查可指示50岁以上患者的慢性肝脏受累和肝脏影像学
- 肾脏. 肾脏功能检查以评估肾脏损害的存在和进展
- 皮肤. 评估皮肤起泡性病变以评估其与VP的关系
- 药物(请参阅要Agents/Circumstances to Avoid),饮食和并发情况对VP严重程度的影响
- 咨询临床遗传学家和/或遗传咨询师
表现治疗
神经内脏症状 大多数急性神经内脏发作需要入院;轻度发作(不需要麻醉止痛药且无低钠血症,癫痫发作或肌肉无力)的患者有时被视为门诊患者。住院管理可以优化快速,全面和多学科的评估。
与其他急性卟啉症一样,评估应包括鉴别加重药物和其他促发因素。有害药物包括巴比妥酸盐,磺胺类抗生素,灰黄霉素,利福平,大多数抗惊厥药,包括苯妥英钠和卡马西平,酒精,麦角生物碱,胃复安和孕激素。应停止使用有害药物[Balwani et al 2017].
癫痫发作,运动神经病和低钠血症提示严重疾病,应在ICU中进行适当的支持治疗。 MRI可发现可逆性脑血管痉挛的证据[Webb et al 2016]。
疼痛和恩丹西酮或有关恶心和呕吐的相关药物通常需要麻醉性镇痛药。吩噻嗪对恶心和精神症状(例如,躁动,幻觉)也有效[Anderson et al 2005, Harper & Wahlin 2007].
轻度发作(不需要麻醉药且无低钠血症,癫痫发作或运动神经病)可通过葡萄糖负荷治疗,但大多数发作应通过静脉内血红素治疗[Anderson et al 2005, Balwani et al 2017].
注意:“ Hemin”是指原卟啉IX的氧化形式,但也是用于急性卟啉症的静脉治疗的血红素制剂的通用术语,例如冻干的血红素(氢氧化血红素)和精氨酸血红素。当将这些血红素制剂静脉内输注时,血红素与循环白蛋白结合成血红素白蛋白。后者被肝细胞吸收并减少肝ALAS1的合成,肝ALAS1是肝脏中血红素合成的速率控制酶。
急性发作患者应仔细监测可能需要通气支持的肌肉无力和呼吸障碍。
低钠血症应缓慢纠正,并使用不会加重卟啉症的药物治疗癫痫。
VP也需要考虑肝移植,在患有严重反复发作且对药物治疗反应不佳的acute intermittent porphyria患者中肝移植已有效[Dowman et al 2012].
通过控制高血压可以在某种程度上预防肾脏疾病的进展。
皮肤表现 如果可以识别和消除诱发因素,则卟啉水平可能降低,光敏性提高。否则,没有有效的方法可以降低卟啉水平。用血红素治疗可能仅在短期内降低卟啉。
患者应穿防护服,并避免阳光直射。
对于疼痛的病灶可能需要使用止痛药,对于合并的感染可能需要使用抗生素。局部类固醇几乎没有益处。
有效治疗porphyria cutanea tarda的具体措施(即放血和小剂量羟氯喹或氯喹)对VP的治疗无效。
预防主要表现
急性发作
- 如果纠正或避免诱发因素,将来急性发作的可能性就较小(请参阅 Agents/Circumstances to Avoid)。
- 促性腺激素释放激素类似物可预防急性卟啉症经前反复发作,包括促性腺激素释放激素类似物[Anderson et al 1990, Schulenburg-Brand et al 2017].
- 每周或每两周一次的血红素输注可以预防频繁的非周期性发作;然而,缺乏公开的经验[Marsden et al 2015].
- Givosiran是一种小的干扰RNA(siRNA)治疗剂,最近已被FDA批准用于治疗急性卟啉症,包括VP。尤其是,每月皮下注射Givosiran可以有效预防频繁发作的发作 [Sardh et al 2019].
光敏皮肤表现。预防VP的皮肤表现需要防晒。避免诱发因素也是有益的。
监测
患有急性卟啉症且胆色素原或卟啉持续升高的患者尤其可能在50岁以后发展为肝细胞癌;从50岁开始每隔六个月进行一次肝脏成像可发现早期病变 [Andant et al 2000, Schneider-Yin et al 2010].
避免的药物/情况
应避免的诱发因素包括:巴比妥酸盐,磺胺类抗生素,灰黄霉素,利福平,大多数抗惊厥药,包括苯妥英和卡马西平,酒精,麦角麦角生物碱,胃复安和孕激素。
更新的列表在 American Porphyria Foundation(美国卟啉症基金会)和 European Porphyria Network(欧洲卟啉症网络)的网站上。
尽管通常应避免使用避孕药,但可以耐受低剂量的激素制剂。
应该避免空腹和非常低卡路里的饮食。 VP和其他急性卟啉症常发作的患者应避免减肥手术。希望减肥的患者应在营养师的指导下逐步进行,并在一定程度上长期减少卡路里摄入量。
评估处于危险亲戚
明确先证者的无症状高危和低危亲戚的遗传状况是适当的。这将确定家族性PPOX 致病性变异的杂合的个体,他们可能会从有关合理使用药物和避免已知诱发因素的咨询中受益。
有关与 遗传咨询目的相关的高危亲戚测试的问题,请参见Genetic Counseling。
孕期管理
混合性卟啉症(VP)妇女通常对妊娠耐受良好;但是,某些有VP的妇女在怀孕期间可能会加剧病情。
Badminton & Deybach [2006] 刊登的报告成功地治疗了几名孕妇在怀孕期间使用血红素(以精氨酸血红素形式)发作VP或其他急性卟啉症发作,而没有不良胎儿作用。他们强调,对于急性卟啉症的治疗,几乎从不建议中断妊娠。
氢氧化血红素(血红素)的使用经验也很有限,但提示在怀孕期间没有不良反应 [Isenschmid et al 1992]。如所提到的(见表现疗法,Neurovisceral Symptoms),当血红素以精氨酸血红素或氢氧化血红素形式给药时,血红素以血红素白蛋白的形式传递到组织中,这些制剂有望具有相似的安全性。
杂合的PPOX 致病性变异的胎儿的预后较好,因为当前的产后管理涉及就适当药物使用和避免已知促发因素向家庭提供咨询。
有关怀孕期间使用药物的更多信息,请参见MotherToBaby。
正在调查的疗法
在美国搜索ClinicalTrials.gov,在EU Clinical Trials Register,以获取有关各种疾病和状况的临床研究信息。注意:可能没有针对该疾病的临床试验。
遗传咨询
遗传咨询是为个人和家庭提供有关遗传疾病的性质,遗传和影响的信息,以帮助他们做出明智的医疗和个人决定的过程。 下一节讨论遗传风险评估以及家族史和基因检测的使用,以阐明家族成员的遗传状况。 本节的目的不是要解决个人可能面临的所有个人,文化或伦理问题,也不能替代遗传专家的咨询。 —编者。
遗传方式
混合性卟啉病(VP)以常染色体显性遗传方式遗传, 外显率降低。
家庭成员的风险
先证者的父母
- 通常,诊断为VP的先证者的一位父母是PPOX致病性变异的杂合的个体,可能有也可能没有症状。
- 很少有被诊断为VP的人患有de novo 致病性变异。
- 建议对具有明显 de novo 致病性变异的先证者的父母进行分子遗传学检测。
- 如果在任一亲本的白细胞DNA中都无法检测到 先证者中发现的致病性变异,则可能的解释包括亲本的先证者中的 de novo 致病变异或亲本中的 胚系嵌合。尽管从理论上讲是可行的,但尚无先证者从具有种系镶嵌性的亲本遗传致病性变异的案例。
- 一些确诊为VP的个体的家族史似乎是阴性的,原因是外显率降低或未能将非特异性症状识别为该疾病。因此,除非已对先证者的父母进行了分子遗传学检测,否则无法确认阴性的家族史。
先证者的同胞。先证者同胞的风险取决于先证者父母的遗传状况:
- 如果在先证者的父母发现了先证者中的PPOX 致病性变异,则每个同胞都有50%的风险遗传该变异。遗传致病变异的同胞可能会或可能不会出现VP症状。
- 如果先证者具有已知的与VP相关的致病性变异,而在任一亲本的白细胞DNA中均无法检测到,则由于亲本 胚系嵌合理论的可能性,估计同胞的 再发风险为1%[Rahbari et al 2016].
先证者的后代
其他家庭成员
相关的遗传咨询问题
有关评估以早期诊断和治疗为目的的高风险亲戚的信息,请参阅《管理,Evaluation of Relatives at Risk 》。
具有明显de novo 致病性变异的家庭的注意事项。当具有常染色体显性遗传条件的先证者的父母均未鉴定出致病变异或无该疾病的临床证据时,该致病变异很可能是新发的,也可以探索非医学解释,包括非生物学父亲 或产妇(例如,辅助生殖)和未公开的收养。
家庭计划
DNA库是DNA(通常从白细胞中提取)的存储,以备将来使用。因为将来测试方法和我们对基因,等位基因变异和疾病的理解可能会有所改善,所以应考虑受累的患者DNA库。
产前检测和植入前基因检测
一旦在一个受累的家庭成员中发现了PPOX致病性变异,就可以对风险增加的妊娠进行产前检查,对VP进行植入前基因检查。注意:通过产前检查检测到的PPOX致病变体的存在并不能预测个体是否有症状,发作年龄或表现形式。
在医疗专业人员之间以及在家庭内部,关于使用产前检查的观点可能存在差异,特别是如果考虑将检查用于终止妊娠而不是早期诊断的时候。尽管大多数中心将有关产前检查的决定视为父母的选择,但对这些问题的讨论是适当的。
资源
GeneReviews工作人员选择了以下特定疾病和/或综合保护组织和/或注册表,以保护患有这种疾病的个人及其家人。 GeneReviews对其他组织提供的信息概不负责。 有关选择标准的信息,请单击 here.
- American Porphyria Foundation (APF)4915 St. Elmo AvenueSuite 200Bethesda MD 20814Phone: 1-866-APF-3635 (toll-free); 301-347-7166Email: porphyrus@porphyriafoundation.com
- British & Irish Porphyria Network (BIPNET)United Kingdom
- British Porphyria AssociationUnited KingdomPhone: 0300 30 200 30Email: helpline@porphyria.org.uk
- Canadian Association for Porphyria/Association Canadienne de PorphyrieCanada
- Find a Porphyria ExpertAmerican Porphyria Foundation
- National Acute Porphyria Service (NAPS)United KingdomPhone: +44 29 2184 7747
- National Library of Medicine Genetics Home Reference
- Porphyria South AfricaSouth AfricaPhone: +27 21-4066332Fax: +27 21-4066061Email: Peter.Meissner@uct.ac.za
- Welsh Medicines Information CentreThe Welsh Medicines Information Centre (WMIC) offers a specialist advisory service on the safe use of drugs in porphyria.United KingdomPhone: +44 029 2074 4298
- European Porphyria NetworkEmail: contact@porphyria.eu
- Swedish Porphyria Patients' AssociationKarolinska UniversitetssjukhusetHuddinge M 96Stockholm Stockholms Lan SE-141 86SwedenPhone: +46 8 711 56 09Email: porfyrisjukdomar@gmail.com
分子遗传
分子遗传学和OMIM表中的信息可能不同于GeneReview中的其他信息:表中可能包含最新信息. —编者.
Table A
混合性卟啉病 :基因和数据库
| 基因 | 染色体定位 | 蛋白 | 数据库 | HGMD | ClinVar |
|---|---|---|---|---|---|
| PPOX | 1q23 | Protoporphyrinogen oxidase | PPOX database | PPOX | PPOX |
Table B
混合性卟啉病的OMIM 条目 (View All in OMIM)
分子发病机理
PPOX催化原卟啉原氧化为原卟啉,并除去六个质子。 VP中PPOX的部分缺乏限制了血红素的合成,特别是在存在导致肝血红素合成增加和在肝脏中诱导δ-氨基乙酰丙酸合酶-1(ALAS1)产生的抑制因素的情况下。 ALAS1是ALAS的一种普遍存在的形式,在所有组织中都可以发现,这与仅在骨髓中产生的红细胞特异性形式的ALAS2相反。
病因的机制。 VP是由 杂合的 功能丧失性 PPOX变异引起的,导致所有组织中的酶减少至正常水平的约50%。
Table 4
值得注意的PPOX致病变异
| 参考序列 | DNA 核酸改变 | 蛋白质改变 | 备注 [Reference] |
|---|---|---|---|
| NM_001122764 NP_001116236 | c.175C>T | p.Arg59Trp | 南非Afrikaner人口的建立者变异在非洲的VP中约占95% [Dean 1971, Meissner et al 1996] |
表中列出的变异由作者提供。 GeneReviews员工尚未独立验证变异的分类。GeneReviews遵循人类基因组变异学会(varnomen
- .hgvs.org ).的标准命名约定。 有关命名法的说明,请参见Quick Reference。
参考文献
引用文献
- Andant C, Puy H, Bogard C, Faivre J, Soulé JC, Nordmann Y, Deybach JC. Hepatocellular carcinoma in patients with acute hepatic porphyria: frequency of occurrence and related factors. J Hepatol. 2000;32:933 - 9. [PubMed: 10898313]
- Anderson KE, Bloomer JR, Bonkovsky HL, Kushner JP, Pierach CA, Pimstone NR, Desnick RJ. Recommendations for the diagnosis and treatment of the acute porphyrias. Ann Intern Med. 2005;142:439 - 50. [PubMed: 15767622]
- Anderson KE, Spitz IM, Bardin CW, Kappas A. A gonadotropin releasing hormone analogue prevents cyclical attacks of porphyria. Arch Intern Med. 1990;150:1469 - 74. [PubMed: 2196028]
- Badminton MN, Deybach JC. Treatment of an acute attack of porphyria during pregnancy. Eur J Neurol. 2006;13:668 - 9. [PubMed: 16796597]
- Balwani M, Wang B, Anderson KE, Bloomer JR, Bissell DM, Bonkovsky HL, Phillips JD, Desnick RJ. Acute hepatic porphyrias: recommendations for evaluation and long-term management. Hepatology. 2017;66:1314 - 1322. [PMC free article: PMC5605422] [PubMed: 28605040]
- Barbaro M, Kotajärvi M, Harper P, Floderus Y. Partial protoporphyrinogen oxidase (PPOX) gene deletions, due to different Alu-mediated mechanisms, identified by MLPA analysis in patients with variegate porphyria. Orphanet J Rare Dis. 2013;8:13. [PMC free article: PMC3554555] [PubMed: 23324528]
- Dean G. The Porphyrias: A Study of Inheritance and Environment. 2 ed. London, UK: Pitman Medical; 1971.
- Dowman JK, Gunson BK, Mirza DF, Bramhall SR, Badminton MN, Newsome PN, et al. Liver transplantation for acute intermittent porphyria is complicated by a high rate of hepatic artery thrombosis. Liver Transpl. 2012;18:195 - 200. [PMC free article: PMC3472026] [PubMed: 21618697]
- Elder G, Pauline Harper P, Michael Badminton M, Sandberg S, Deybach J-C. The incidence of inherited porphyrias in Europe. J Inherit Metab Dis. 2013;36:849 - 57. [PubMed: 23114748]
- Frank J, McGrath J, Lam H, Graham RM, Hawk JL, Christiano AM. Homozygous variegate porphyria: identification of mutations on both alleles of the protoporphyrinogen oxidase gene in a severely affected proband. J Invest Dermatol. 1998;110:452 - 5. [PubMed: 9540991]
- Harper P, Wahlin S. Treatment options in acute porphyria, porphyria cutanea tarda, and erythropoietic protoporphyria. Curr Treat Options Gastroenterol. 2007;10:444 - 55. [PubMed: 18221605]
- Hift RJ, Meissner PN. An analysis of 112 acute porphyric attacks in Cape Town, South Africa: Evidence that acute intermittent porphyria and variegate porphyria differ in susceptibility and severity. Medicine (Baltimore). 2005;84:48 - 60. [PubMed: 15643299]
- Isenschmid M, König C, Fässli C, Haenel A, Hänggi W, Schneider H. Acute intermittent porphyria in pregnancy: glucose or hematin therapy? Schweiz Med Wochenschr. 1992;122:1741 - 5. [PubMed: 1448679]
- Kauppinen R, Mustajoki P. Prognosis of acute porphyria: occurrence of acute attacks, precipitating factors, and associated diseases. Medicine (Baltimore). 1992;71:1 - 13. [PubMed: 1549056]
- Marsden JT, Guppy S, Stein P, Cox TM, Badminton M, Gardiner T, Barth JH, Stewart MF, Rees DC. Audit of the use of regular haem arginate Infusions in patients with acute porphyria to prevent recurrent symptoms. JIMD Reports. 2015;22:57 - 65. [PMC free article: PMC4486272] [PubMed: 25762493]
- Mauzerall D, Granick S. The occurrence and determination of δ-aminolevulinic acid and porphobilinogen in urine. J Biol Chem. 1956;219:435 - 46. [PubMed: 13295297]
- Meissner P, Hift RJ, Corrigall A. Variegate porphyria. In: Kadish KM, Smith K, Guilard R, eds. Porphyrin Handbook, Part II. Vol 14. San Diego, CA: Academic Press; 2003:93-120.
- Meissner PN, Dailey TA, Hift RJ, Ziman M, Corrigall AV, Roberts AG, Meissner DM, Kirsch RE, Dailey HA. A. R59W mutation in human protoporphyrinogen oxidase results in decreased enzyme activity and is prevalent in South Africans with variegate porphyria. Nat Genet. 1996;13:95 - 7. [PubMed: 8673113]
- Peters T. King George III, bipolar disorder, porphyria and lessons for historians. Clin Med (Lond). 2011;11:261 - 4. [PMC free article: PMC4953321] [PubMed: 21902081]
- Pinder VA, Holden ST, Deshpande C, Siddiqui A, Mellerio JE, Wraige E, Powell AM. Homozygous variegate porphyria presenting with developmental and language delay in childhood. Clin Exp Dermatol. 2013;38:737 - 40. [PubMed: 24073655]
- Poh-Fitzpatrick MB. A plasma porphyrin fluorescence marker for variegate porphyria. Arch Dermatol. 1980;116:543 - 7. [PubMed: 7377785]
- Rahbari R, Wuster A, Lindsay SJ, Hardwick RJ, Alexandrov LB, Turki SA, Dominiczak A, Morris A, Porteous D, Smith B, Stratton MR, Hurles ME, et al. Timing, rates and spectra of human germline mutation. Nat Genet. 2016;48:126 - 33. [PMC free article: PMC4731925] [PubMed: 26656846]
- Sardh E, Harper P, Balwani B, Stein P, Rees D, Bissell DM, Desnick R, Parker C, Phillips J, Bonkovsky HL, Vassiliou D, Penz C, Chan-Daniels A, He Q, Querbes W, Fitzgerald K, Kim JB. MD, Garg P, Vaishnaw A, Simon AR, Anderson KE, A phase 1 study of RNA interference therapy for acute intermittent porphyria. NEJM. 2019;380:549 - 58. [PubMed: 30726693]
- Schneider-Yin X, van Tuyll van Serooskerken AM, Went P, Tyblewski W, Poblete-Gutiérrez P, Minder EI, Frank J. Hepatocellular carcinoma in variegate porphyria: a serious complication. Acta Derm Venereol. 2010;90:512 - 5. [PubMed: 20814629]
- Schulenburg-Brand D, Gardiner T, Guppy S, Rees DC, Stein P, Barth J, Stewart MF, Badminton M. An audit of the use of gonadorelin analogues to prevent recurrent acute symptoms in patients with acute porphyria in the United Kingdom. JIMD Reports. 2017;36:99 - 107. [PMC free article: PMC5680288] [PubMed: 28220410]
- van Tuyll van Serooskerken AM, de Rooij FW, Edixhoven A, Bladergroen RS, Baron JM, Joussen S, Merk HF, Steijlen PM, Poblete-Gutiérrez P, te Velde K, Wilson JH, Koole RH, van Geel M, Frank J. Digenic inheritance of mutations in the coproporphyrinogen oxidase and protoporphyrinogen oxidase genes in a unique type of porphyria. J Invest Dermatol. 2011;131:2249 - 54. [PubMed: 21734717]
- Webb AJS, Ingale H, Irani SR, Hu MTM. Acute variegate porphyria presenting with reversible cerebral vasoconstriction. Clin Neurol Neurosurg. 2016;146:102 - 4. [PubMed: 27186968]
- Whatley SD, Mason NG, Woolf JR, Newcombe RG, Elder GH, Badminton MN. Diagnostic strategies for autosomal dominant acute porphyrias: retrospective analysis of 467 unrelated patients referred for mutational analysis of the HMBS, CPOX, or PPOX gene. Clin Chem. 2009;55:1406 - 14. [PubMed: 19460837]
- Whatley SD, Puy H, Morgan RR, Robreau AM, Roberts AG, Nordmann Y, Elder GH, Deybach JC. Variegate porphyria in Western Europe: identification of PPOX gene mutations in 104 families, extent of allelic heterogeneity, and absence of correlation between phenotype and type of mutation. Am J Hum Genet. 1999;65:984 - 94. [PMC free article: PMC1288269] [PubMed: 10486317]
本章节备注
致谢
这项工作得到了NIH / NIDDK和罕见疾病临床研究网络的一部分资助的卟啉症联盟以及美国卟啉症基金会的支持。
更新历史
- 12 December 2019 (bp) 系统性更新发布到公开网页上
- 14 February 2013 (me) 综述内容发布到公开网页上
- 12 June 2012 (kea) 初稿提交