
概述
临床特点.
REN相关 常染色体显性肾小管间质性肾病 ( Autosomal dominant tubulointerstitial kidney disease, REN-related ,ADTKD- REN) (以前称为 家族性青少年型高尿酸血症性肾病2型[ 家族性 juvenile hyperuricemic nephropathy type 2,FJHN2]) 的特征是:绝大多数 受累儿童在一岁之前即可发现低增生性贫血伴血红蛋白浓度降低;绝大多数(不是全部)受累个体可见高尿酸血症和痛风;以及缓慢进展的慢性肾小管间质性肾病。一些受累儿童有多尿症(尿量过多导致尿频)和 遗尿。无症状儿童的血清肌酐浓度轻度升高和肾小球滤过率下降,常常在40至60岁时,进展为终末期肾病( end-stage renal disease,ESRD)。值得注意的是,仅14 个存在这种情况家系的信息披露;如有更多的家系报道,可能对此病有更好的了解。
诊断/检测.
诊断依据低增生性贫血(相对于血红蛋白浓度,网织红细胞计数低,并且促红细胞生成素浓度低),高尿酸血症(血清尿酸浓度>6mg/dL),尿尿酸排泄分数降低,尿沉渣无特殊和低血浆肾素活性。肾脏大小正常或缩小,无囊肿形成的证据。 REN(编码肾素) 是唯一已知的突变可导致ADTKD- REN的 基因。
管理.
对症治疗:促红细胞生成素也许可纠正贫血;急性痛风通常对强的松和秋水仙碱反应良好。低血浆肾素活性/低血浆醛固酮浓度的 治疗应在进展至慢性肾病III期之前。如果有高钾血症,应予氟氢可的松或限钾治疗。当发生终末期肾病时,可行肾移植。 移植肾不会复发ADTKD- REN,它可以正常地产生肾素。
预防主要临床症状:别嘌醇治疗高尿酸血症可预防痛风。
监管:从诊断时开始,每年测血红蛋白浓度、血清尿酸和肌酐浓度。
需避免的药物/环境:避免使用非甾体类抗炎药,尤其是脱水者。血管紧张素转化酶抑制剂可能加重潜在的相对肾素缺乏。血容不足、脱水、高肉类和海产品摄入可能加重痛风。患者不应低钠饮食(其通常用于慢性肾病)。
亲属风险评估:如果一个 受累家系成员的 REN致病性变异 已被确定,高危亲属可以考虑做 分子遗传学检测,特别是儿童(因为他们患贫血的风险高)、青少年(因为他们患痛风的风险高,而这是可以通过适当治疗来预防的)和考虑捐献肾脏的亲属。
诊断
提示性发现
有下列临床表现的个体,应考虑 REN相关 常染色体显性肾小管间质性肾病 ( Autosomal dominant tubulointerstitial kidney disease, REN-related,ADTKD- REN):
- 低增生性贫血伴血红蛋白浓度降低,由于促红细胞生成素低,见于绝大多数一岁以内的 受累儿童。
- 血红蛋白浓度可降低(通常为 9-11g/dL)。
- 相对于血红蛋白浓度,网织红细胞计数降低。
- 促红细胞生成素浓度低。
- 血液学所有其它方面均正常。
- 高尿酸血症和痛风,由于肾脏排泄尿酸减少,绝大多数(不是全部) 患病个体可见高尿酸血症和痛风。
- 80%的 受累个体在儿童时期开始有高尿酸血症(血清尿酸浓度>6mg/dL)。
- 通常,高尿酸血症在肾功能正常的个体中,血清尿酸浓度>1SD的相应年龄和性别的正常值 (见 表 1) [ Wilcox 1996].
- 绝大多数 ADTKD- REN个体尿尿酸排泄分数下降(见 Table 2).
- 缓慢进展的慢性肾小管间质性肾病
- 尿液分析显示尿沉渣无特殊(即几乎无血细胞和蛋白)。通常无血尿,并且蛋白质排泄<1 g/24h,除非进展为肾衰竭。
- 血浆肾素低。
- 血浆醛固酮低。
- 血清钾轻度升高。
- 肾脏超声显示肾脏大小正常或偏小,无囊肿。
- 肾活检(见注):
组织学检查显示局灶性肾小管萎缩,继发性肾小球瘢痕形成和间质纤维化 [ Zivná et al 2009]。
与对照组织相比,肾小球旁器的颗粒细胞中,肾上腺素和肾素原的免疫染色显著降低(其在疾病早期检测不到)。在进展期,肾小球旁器的颗粒细胞和肾小管上皮细胞均无肾上腺素和肾素染色。
注:不应行肾活检来诊断ADTKD- REN,因为肾活检是有一定风险的侵入性操作,并且病理学发现无特异性,不能据此明确病因。分子遗传学检测(见 建立诊断)是诊断的金标准,比肾活检更安全、便宜。然而, 一些 受累个体(或其亲属)可能在考虑诊断ADTKD- REN可能之前,已行肾活检。
Table 1.
肾功能正常个体血清尿酸浓度
| 年龄 | 血清浓度 (mg/dL) | |
|---|---|---|
| 男性 | 女性 | |
| <5 岁 | 3.6±0.9 | 3.6±0.9 |
| 5-10岁 | 4.1±1.0 | 4.1±1.0 |
| 12岁 | 4.4±1.1 | 4.5±0.9 |
| 15岁 | 5.6±1.1 | 4.5±0.9 |
| >18 岁 | 6.2±0.8 | 4.0±0.7 |
Table 2.
肾功能正常个体尿尿酸排泄分数
| 年龄 | 平均 | 标准差 1 | |
|---|---|---|---|
| 0-6周 | 29.1% | 11.7 | |
| 6周 - 1岁 | 23.9% | 10.4 | |
| 1-3岁 | 15.2% | 6.2 | |
| 3-13岁 | 12.2% | 5.5 | |
| >13岁 | 女性 | 8.0% | 3.7 |
| 男性 | 10.3% | 4.2 | |
建立诊断
ADTKD- REN的诊断是 基于先证者通过 分子遗传学检测在 REN基因中发现了 杂合致病性变异 (见 Table 3)。
分子检测方法包括 单基因检测,用多基因 panel,和 更全面的 基因组检测。
Table 3.
用于 REN相关常染色体显性肾小管间质性肾病(ADTKD- REN)的分子遗传检测总结
临床特点
临床描述
目前仅14个家系已经确定患有 REN相关 常染色体显性肾小管间质性肾病( autosomal dominant tubulointerstitial kidneydisease,ADTKD- REN);因此,临床表现谱可能尚未充分认识。
10岁前。如日常照顾良好,ADTKD- REN儿童可在1岁时就因发现血红蛋白浓度降低而诊断。虽然儿童无症状,但是这时肾功能检查可有轻度肾功能不全。
受累但尚未确诊的儿童,在因发热性疾病使用非甾体类抗炎药( nonsteroidal anti-inflammatory drug ,NSAID)时, 可能因为低血浆肾素活性、血容不足和前列腺素抑制的联合作用,导致急性肾脏衰竭[ Bleyer et al 2010a]。如果治疗得当,急性肾衰竭通常可以治愈,但是慢性肾脏衰竭和贫血可能在那时开始被发现。
伴随低尿酸排泄分数的高尿酸血症出现在儿童时期,但是通常没有症状。
10-20岁。随着从儿童时期进入到青春期,贫血消失,血红蛋白浓度上升到正常范围。
尿酸浓度升高可能导致痛风。痛风与大脚趾、前脚、脚后跟、膝盖和其它关节的疼痛和红斑急性发作有关。痛风因为可能发生在儿童或年轻女性中(传统的痛风发生于中年男性),而常常漏诊。痛风应迅速予强的松或秋水仙碱治疗。长期服用别嘌醇可预防痛风。
一些 受累个体(不是全部)有:
- 由于尿浓缩功能降低,导致多尿症和尿量过多[ Bleyer et al 2010a]。 多尿症出现在儿童时期,一直延续到成年期。
- 轻度低血压;
- 血钾轻度升高。
成年。肾功能随时间慢慢恶化。无症状儿童的轻度血肌酐升高和肾小球滤过率降低,通常在40至60岁时进展为终末期肾病( end-stage renal disease,ESRD)。在40到70岁时,可能需要肾脏替代疗法。肾移植可以治愈本病。
命名法
根据2015的命名法[ Eckardt et al 2015],术语 “ 常染色体显性肾小管间质性肾病” ( autosomal dominanttubulointerstitial kidney disease,ADTKD) 是指以下特征的疾病:(1)常染色体显性遗传;(2)缓慢进展的慢性肾小管间质性肾病,在30至70岁导致ESRD;(3)尿液分析显示尿沉渣无特殊(即几乎无血细胞和蛋白);(4)疾病早期阶段,肾脏超声检查正常[ Bleyer et al 2010a]:
- MUC1相关 常染色体显性肾小管间质性肾病( Autosomal dominant tubulointerstitial kidney disease – MUC1-related,ADTKD- MUC1;以前称为 髓质囊性肾病1型)[ Kirby et al 2013]
- UMOD相关 常染色体显性肾小管间质性肾病( Autosomal dominant tubulointerstitial kidney disease – UMOD-related,ADTKD- UMOD;以前称为 UMOD-相关肾脏疾病, 家族性青少年型高尿酸血症性肾病1型, 髓质囊性肾病2型[MCKD2],或尿调制素贮积病)
- REN相关 常染色体显性肾小管间质性肾病( Autosomal dominant tubulointerstitial kidney disease – REN-related,ADTKD- REN;以前称为 家族性青少年型高尿酸血症性肾病2型或 REN相关肾脏疾病[ Zivná et al 2009]
注:(1)过去使用术语“肾消耗病/髓质囊性肾病( nephronophthisis/medullary cystic kidney disease,NPH/MCKD)复合体” 是指 常染色体隐性和 常染色体显性两种形式的遗传性慢性肾小管间质疾病[ Hildebrandt et al 1992]。肾消耗病现在是指常染色体隐性遗传的一组在儿童时期出现慢性肾脏衰竭的疾病。这些疾病可由至少12个不同基因的致病变异引起,指nephrocystins( NPHP1- NPHP11, NPHP1L)[ Wolf & Hildebrandt 2011]。临床特点包括多尿症、贫血和慢性进行性肾脏衰竭。见 肾消耗病。(2)与肾脏髓质钙化、高尿钙、血尿和肾小管酸化缺陷有关的髓质海绵肾( medullary sponge kidney,MSK)[ Gambaro et al 2006],与髓质囊性肾病无关。
患病率
REN-相关 常染色体显性肾小管间质性肾病( Autosomal dominant tubulointerstitial kidney disease, REN-related,ADTKD- REN)非常罕见,全世界仅确认了约14个家系。一些研究人员研究了大量患有遗传性肾脏疾病的家系,只有少数几个家系确认有此病。
遗传相关(等位基因)疾病
除了在此 GeneReview中讨论的那些表型之外,没有已知的其它表型与 REN基因 杂合致病性变异有关。
功能丧失( 无效)的纯合或复合杂合 REN 变异导致大体正常的肾脏出现近端肾小管分化丧失。常染色体隐性肾小管发育不全在围产期是致死的[ Gribouval et al 2005]。
鉴别诊断
图 1提供了帮助遗传性肾脏疾病鉴别诊断的流程图。
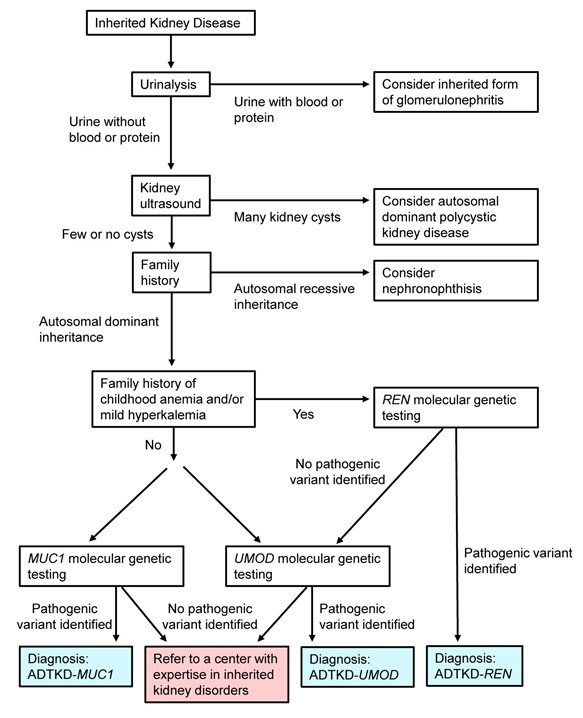
图 1.
遗传性肾脏疾病的检测策略–2015年更新
遗传性肾小球肾炎。受累个体通常有蛋白尿和/或血尿。如果尿液中存在血细胞或蛋白,考虑遗传性肾小球肾炎(如: Alport 综合征)。 尽管极少数 ADTKD- UMOD个体发现有蛋白尿,但这是不典型的。
常染色体显性多囊肾(autosomal dominant polycystic kidney disease ,ADPKD)。如果 常染色体显性遗传方式的肾脏疾病患者尿沉渣无特殊(即几乎无血细胞和蛋白),则必须排除ADPKD,大于25岁的 受累个体肾脏超声检查可见大量囊肿。
其他形式的ADTKD。如果个体没有ADPKD且尿沉渣无特殊,则需考虑以下另外两种形式的 常染色体显性肾小管间质性肾病:
- ADTKD- MUC1与 MUC1致病性变异有关。受累个体缓慢进展的慢性肾病和微量蛋白尿。一个重要的区别因素是ADTKD- MUC1痛风通常仅发生于III期或以后的慢性肾病。受累个体在儿童时期没有贫血和其它表现。
- ADTKD -UMOD。与ADTKD- REN一样,这种疾病与缓慢进展的慢性肾病有关。 UMOD相关的肾病患者在儿童时期没有贫血,并且没有在ADTKD- REN中经常见到的轻度高钾血症。
肾消耗病(Nephronophthisis, NPH)是一种 常染色体隐性肾小管间质性肾病。与ADTKD- REN一样,通常见于儿童时期,并且与贫血和轻度低血压有关。这两种疾病在以下三个方面不同:(1)ADTKD- REN通常与肾脏疾病、贫血和痛风的家族史相关。在肾消耗病中,通常没有阳性的家族史,因为其以常染色体隐性方式遗传。(2)ADTKD- REN在婴儿期即出现贫血,而在肾消耗病中,贫血通常与肾功能水平相关。(3)肾脏衰竭通常在肾消耗病中更严重, 受累个体通常在十几岁和二十几岁时需要透析治疗。相比之下,ADTKD- REN个体到成年早期,eGFR通常>50 mL/min。NPH与至少有19种已知基因的致病性变异相关。
Fabry病,一种 X-连锁疾 病,由α-半乳糖苷酶(α-Gal)A活性缺引起,酰基鞘鞍醇三己糖(GL-3)进行性沉积于全身细胞的溶酶体中。经典型发生于α-Gal A活性<1%的男性,通常在儿童或青春期起病,表现为四肢(肢端感觉异常)周期性发作的剧烈疼痛、血管性皮肤病变(血管角质瘤)、少汗症、特征性角膜和晶 状体浑浊,以及蛋白尿(通常超过 ADTKD- UMOD中见到的蛋白尿)。通常在30至50岁时肾功能逐渐恶化为终末期肾病(ESRD)。α-Gal A活性> 1% 的男性有心脏或肾脏变异 表型。罕见的是, 杂合的女性 携带者可能出现有与经典型男性一样严重的症状。
管理
初步诊断后的评估
为了确定疾病的程度和个体诊断为ADTKD- REN的需要, 推荐进行以下评估:
- 血红蛋白浓度以记录贫血水平
- 血清尿酸浓度以确定有痛风风险者
- 收集24小时尿以定量尿量,并确定是否存在多尿症(虽然这通常可以通过历史记录来确定)。问诊关于遗尿(尿床)、频繁口渴或排尿的信息将有助于建立多尿症的诊断。
- 标准的基本代谢全套检查来确定是否存在高钾血症
- 咨询临床遗传学家或遗传咨询师
对症处理
贫血可以用促红细胞生成素治疗逆转[ Zivná et al 2009], 用药方式为皮下注射,由血液科医生或儿科肾脏病医生管理。药物使用剂量需根据治疗的反应进行调节,目前没有明确的目标,可由血液科医生或肾脏病医生灵活决定。许多儿 童有轻微的贫血,血红蛋白水平为10至11g/dL,并且不使用促红细胞生成素也是安全的。
注:随着血红蛋白浓度在青春期增加,促红细胞生成素的剂量需要降低。
高尿酸血症/痛风。痛风通常对强的松和秋水仙碱反应良好。ADTKD- REN个体不应使用非甾体类药物,因为非甾体类药物和ADTKD- REN患者的低肾素状态联合作用,可能导致急性肾脏损伤;强的松是更好的选择。
患痛风的个体应考虑用别嘌醇或丙磺舒来预防未来的痛风发作。经别嘌醇治疗,血清尿酸浓度可恢复正常,完全防止痛风发作。可能需要终身别嘌醇治疗。 过敏或不耐受别嘌醇个体,可以考虑非布索坦;然而,没有这种药物在 REN相关肾病中使用的资料。
低血压和轻度高钾血症可能出现在患本病的儿童和青壮年中。随着慢性肾病进展至慢性肾病III期,可出现高血压,而且高钾血症不是由于低血浆肾素活性引起,而是肾脏排钾减少所致。
因此,低血浆肾素活性/低血浆醛固酮浓度的 治疗应在进展至慢性肾病III期之前。如果患者有轻度的低血压和高钾血症但保留了肾脏功能,这样的治疗可能包括自由钠的摄入。3-4 g/天的膳食钠摄入量可能预防低血压。注:ADTKD- REN患者不应低钠饮食。
如果存在高钾血症,应予氟氢可的松或限钾治疗[ Bleyer et al 2010b]。低血浆肾素活性的 受累儿童用氟氢可的松治疗(0.1 mg 口服/天)可使血压轻度升高、纠正轻度高钾血症和肾小球滤过率显著改善(可能为血流动力学介导)[ Bleyer at al 2010b]。使用氟氢可的松还可预防可能因血容不足引起的低血压,例如与病毒综合征相关或剧烈运动后过度出汗所致的血容不足。注:氟氢可的松治疗两个晚期肾病的成年人,未见临床效果[作者,个人观察]。
氟氢可的松有助于减少肾素产生(通过负反馈),从而减少异常肾素沉积物的生成。然而,由于阻断肾素血管紧张素系统和醛固酮产生是慢性肾病的 一般疗法,醛固酮的血清浓度增加可能加快肾病的进展。因此,虽然本病使用氟氢可的松可能是有益的,但目前其使用还是由临床医生决定。
盐皮质激素替代。由于患本病的个体很少并且治疗 受累个体的经验有限,关于治疗的建议没有证据基础。
肾脏疾病。建议转诊到肾脏科医生以监测肾功能,评估慢性肾病的表现,当出现肾功能不全时准备肾脏替代治疗。
肾脏替代治疗如血液透析和腹膜透析代替肾脏功能,但与潜在的并发症相关。
肾脏移植治愈ADTKD- REN。移植肾不会发生本病。
预防并发症
如果存在铁缺乏(一个无关的疾病),应根据需要补充铁储存。
监管
在疾病诊断时开始,每年检测血红蛋白浓度、血清尿酸浓度和血清肌酐浓度。
需避免的药物/环境
应避免使用非甾体类抗炎药( nonsteroidal anti-inflammatory medications,NSAIDs),尤其是脱水的患者。患有ADTKD- REN的发热儿童使用NSAIDs可能导致急性肾衰竭[ Bleyer et al 2010b]。应考虑使用其它镇痛药/解热药。
使用血管紧张素转化酶抑制剂在治疗慢性肾衰中可能没有益处,并且可能加重潜在的相关肾素缺乏。
血容不足和脱水可能加重高尿酸血症,并导致更频繁的痛风发作。
高肉类和海产品的摄入可能加剧痛风。
应该避免在极端条件下(例如:天气热时进行强体力活动)的运动。
亲属风险评估
通过对 家族性REN致病性变异的 分子遗传学检测来评估 受累个体的似乎无症状亲属是适当的,以便尽早发现那些将从起始治疗和预防措施中获益者。特别重要的是:
- 儿童:因为他们患贫血的风险高;
- 青少年:因为他们患痛风的风险高,这可用别嘌醇治疗预防;
- 有意捐赠肾脏给 受累家系成员的亲属。
妊娠管理
已有携带 REN致病性变异的妇女成功怀孕的文献记载,流产或其他不良结果的概率没有增加。
还在研究阶段的治疗方法
搜索 ClinicalTrials.gov以获取大量疾病的临床研究信息。注:本病可能没有临床试验。
遗传咨询
遗传咨询是向患者及其家庭提供遗传病的性质、遗传方式及其影响的信息,以帮助他们做出明智的医疗和个人决定的过程。接下来的部分涉及遗传风险评估,根据家族史和遗传学检测确认家庭成员的遗传状态。本节不是为了解决个体可能面临的所有个人、文化或伦理问题,或替代遗传学专业人士的咨询—ED.
遗传模式
REN相关 常染色体显性肾小管间质性肾病 (ADTKD- REN)以 常染色体显性模式遗传。
家系成员的风险
先证者的父母
- 大多数诊断为ADTKD- REN的个体都有一位 受累的父(母)亲。
- 父母的评估可能确定一个 受累个体的诊断,其以前因保健专业人员未能识别该疾病和/或因为表型轻微而未能诊断。
- 父母通常在儿童时期患有贫血,但他们可能不记得和/或者当时可能未能诊断贫血。
先证者的同胞。先证者同胞的风险取决于先证者父母的遗传状况:
- 当父母临床上未受累时, 先证者同胞的风险低。
先证者的后代。ADTKD- REN个体的每个孩子都有50% 的机会获得 REN 致病性变异。
其他家系成员。其他家系成员的风险取决于 先证者父母的遗传状况:如果父母存在 REN致病性变异,他或她的家系成员可能有风险。
遗传咨询的相关问题
为了早期诊断和治疗,评估高危亲属的相关信息,参见管理、 亲属风险评估。
表面上新发致病性变异家系的思考。当 先证者父母双方均未患ADTKD- REN时, REN的致病变异可能是 新发的,然而,需考虑可能的非医学解释包括 替代父亲或产妇(例如辅助生殖)或为秘密收养。
生育规划
- 怀孕前是确定遗传风险和讨论产前检查可用性的最佳时间。
DNA文库是DNA(通常从白细胞中提取)的储存所,以备将来使用。因为测试方法和我们对基因、等位基因变异和疾病的理解可能会在将来得到改善,所以应考虑存储 受累个体的DNA。
产前检测和胚胎植入前的遗传学诊断
一旦在 受累的家系成员中确认了 REN致病变异,可选择对有患ADTKD- REN 风险的胎儿行产前检测和 胚胎植入前的遗传学诊断。
在医学专业人员和家庭内部成员之间,关于产前检测可能存在分歧,特别是如果检测的目的是为了终止妊娠而不是早期诊断。虽然大多数中心会认为关于产前检查的决定是父母的选择,但应讨论这些问题。
信息来源
为了该病患者及其家庭成员的方便,GeneReviews的员工已经选择了下述的针对本病或者包括其他 GeneReviews疾病的患者支持组织和患者注册组织。GeneReviews不为其他组织提供的信息承担责任。选择这些组织的标准,请点击这里获取详细信息。
- REN-相关肾脏疾病登记处Dr. Anthony Bleyer 建立了一个REN突变个体登记处。如果有兴趣参与,请联系他(ableyer@wfubmc.edu)。Email:ableyer@wfubmc.edu
分子遗传学
在下面的分子生物学表和OMIM表内的信息可能和GeneReview里其它部分的信息不一致:表格里的信息可能更加新—ED。
Table B.
常染色体显性肾小管间质性肾病,REN-相关的OMIM入口 ( View All in OMIM)
分子遗传发病机制
肾素是一种由前肾素原生成的天冬氨酰蛋白酶,含有指导内质网(ER)靶向、糖基化和蛋白水解加工的信号序列[ Imai et al 1983]。肾素将血管紧张素原水解为血管紧张素,随后刺激醛固酮的产生。已经发现肾素-血管紧张素系统(RAS)具有广泛而多样的作用,包括调节血管张力、肾脏钠钾调控、红细胞生成、渴感、心脏肥大,并在许多器官通过局部的RAS系统起作用[ Paul et al 2006]。
体外研究表明:突变的信号肽的存在影响肾素靶向性,以及前肾素原到ER的共翻译 易位, 并因此影响肾素原的生物合成和细胞内运输。这导致ER应激、异常的非糖基化前肾素原在胞内积累、加速自噬作用,以及降低生长速率。在体内,这可以使产生 肾素的球旁细胞的活力逐渐降低,并导致肾小管萎缩、肾单位损失和慢性肾衰竭(机制尚不明确)。这类似于在切除了球旁细胞的小鼠身上观察到的现象[ Pentz et al 2004]。
REN致病性变异还可导致肾素生成减少,这是许多慢性肾病的治疗目标。因此,这种疾病成为其自身的治疗。
基因结构。REN位于1号 染色体( 基因组位置 chr1:204123944-204135465;根据 GRCh37/hg19 [Feb 2009] assembly)。该 基因包含10个外显子,其被转录为1493bp的转录本,编码406个氨基酸的前肾素原(参考序列 NM_000537.3)。关于基因和蛋白质信息的详细概述,参见 表 A。
良性的等位基因变异。蛋白C-末端部分的变异与疾病无关,并假定为良性 [ Villard et al 1994; Bleyer & Kmoch, 未公开发表的数据]。
致病性等位基因变异。仅影响肾素生物合成和运输的致病性 错义变异和/或小片段插入/缺失预计将导致ADTKD- REN。1和2号外显子(编码信号肽和前肽的外显子)的致病性变异,预计占导致ADTKD- REN的致病性变异的大多数。
正常的 基因产物。 肾素通常是由肾脏致密斑中入球小动脉经转化的球旁细胞产生。肾素水解切割血管紧张素原N-末端的10个氨基酸,形成血管紧张素I,血管紧张素I通过血管紧张素 转化酶进一步转化为血管紧张素II。血管紧张素II刺激醛固酮的产生。通过多种机制,肾素和血管紧张素有助于维持正常血压并将血清钾浓度降低到正常范围。
异常的基因产物。ADTKD- REN由 REN基因的致病性变异导致,其致病性变异主要位于肾素蛋白的信号序列,影响前肾素原进入ER的靶向性和共翻译 易位。这导致 受累个体的肾活检标本中肾素和肾素-血管紧张素系统的其它组分的低表达。很可能突变蛋白的表达具有 显性毒性效应,逐渐降低肾素表达细胞的活力。这改变了肾内肾素-血管紧张素系统和肾小球旁器的功能,导致肾单位丢失和进行性肾衰竭。
参考资料
参考文献
- Beck BB, Trachtman H, Gitman M, Miller I, Sayer JA, Pannes A, Baasner A, Hildebrandt F, Wolf MT. Autosomal dominant mutation in the signal peptide of renin in a kindred with anemia, hyperuricemia, and CKD. Am J Kidney Dis.2011; 58:821–5.[ PMC free article : PMC3366501] [ PubMed : 21903317]
- Bleyer AJ, Hart PS, Kmoch S. Hereditary interstitial kidney disease. Semin Nephrol.2010a; 30:366–73.[ PMC free article : PMC4264385] [ PubMed : 20807609]
- Bleyer AJ, Zivná M, Hulková H, Hodanová K, Vyletal P, Sikora J, Zivny J, Sovová J, Hart TC, Adams JN, Elleder M, Kapp K, Haws R, Cornell LD, Kmoch S, Hart PS. Clinical and molecular characterization of a family with a dominant renin gene mutation and response to treatment with fludrocortisone. Clin Nephrol.2010b; 74:411–22.[ PMC free article : PMC4264543] [ PubMed : 21084044]
- Eckardt KU, Alper SL, Antignac C, Bleyer AJ, Chauveau D, Dahan K, Deltas C, Hosking A, Kmoch S, Rampoldi L, Wiesener M, Wolf MT, Devuyst O. Autosomal dominant tubulointerstitial kidney disease: diagnosis, classification, and management--A KDIGO consensus report. Kidney Int.2015; 88:676–83.[ PubMed : 25738250]
- Gambaro G, Feltrin GP, Lupo A, Bonfante L, D’Angelo A, Antonello A. Medullary sponge kidney (Lenarduzzi-Cacchi-Ricci disease): a Padua Medical School discovery in the 1930’s. Kidney Int.2006; 69:663–70.[ PubMed : 16395272]
- Gribouval O, Gonzales M, Neuhaus T, Aziza J, Bieth E, Laurent N, Bouton JM, Feuillet F, Makni S, Ben Amar H, Laube G, Delezoide AL, Bouvier R, Dijoud F, Ollagnon-Roman E, Roume J, Joubert M, Antignac C, Gubler MC. Mutations in genes in the renin-angiotensin system are associated with autosomal recessive renal tubular dysgenesis. Nat Genet.2005; 37:964–8.[ PubMed : 16116425]
- Harkness RA, Nicol AD. Plasma uric acid levels in children. Arch Dis Child.1969; 44:773–8.[ PMC free article : PMC2020338] [ PubMed : 5356987]
- Hildebrandt F, Waldherr R, Kutt R, Brandis M. The nephronophthisis complex: clinical and genetic aspects. Clin Investig.1992; 70:802–8.[ PubMed : 1450635]
- Imai T, Miyazaki H, Hirose S, Hori H, Hayashi T, Kageyama R, Ohkubo H, Nakanishi S, Murakami K. Cloning and sequence analysis of cDNA for human renin precursor. Proc Natl Acad Sci U S A.1983; 80:7405–9.[ PMC free article : PMC389959] [ PubMed : 6324167]
- Kirby A, Gnirke A, Jaffe DB, Barešová V, Pochet N, Blumenstiel B, Ye C, Aird D, Stevens C, Robinson JT, Cabili MN, Gat-Viks I, Kelliher E, Daza R, DeFelice M, Hůlková H, Sovová J, Vylet'al P, Antignac C, Guttman M, Handsaker RE, Perrin D, Steelman S, Sigurdsson S, Scheinman SJ, Sougnez C, Cibulskis K, Parkin M, Green T, Rossin E, Zody MC, Xavier RJ, Pollak MR, Alper SL, Lindblad-Toh K, Gabriel S, Hart PS, Regev A, Nusbaum C, Kmoch S, Bleyer AJ, Lander ES, Daly MJ. Mutations causing medullary cystic kidney disease type 1 lie in a large VNTR in MUC1 missed by massively parallel sequencing. Nat Genet.2013; 45:299–303.[ PMC free article : PMC3901305] [ PubMed : 23396133]
- Mikkelsen WM, Dodge HJ, Valkenburg H. The distribution of serum uric acid values in a population unselected as to gout or hyperuricemia: Tecumseh, Michigan 1959-1960. Am J Med.1965; 39:242–51.[ PubMed : 14320691]
- Paul M, Poyan Mehr A, Kreutz R. Physiology of local renin-angiotensin systems. Physiol Rev.2006; 86:747–803.[ PubMed : 16816138]
- Pentz ES, Moyano MA, Thornhill BA, Sequeira Lopez ML, Gomez RA. Ablation of renin-expressing juxtaglomerular cells results in a distinct kidney phenotype. Am J Physiol Regul Integr Comp Physiol.2004; 286:R474–83.[ PubMed : 14563659]
- Stibůrková B, Pospísilová E, Kmoch S, Sebesta I. Analysis of excretion fraction of uric acid. Nucleosides Nucleotides Nucleic Acids.2006; 25:1301–4.[ PubMed : 17065111]
- Villard E, Lalau JD, van Hooft IS, Derkx FH, Houot AM, Pinet F, Corvol P, Soubrier F. A mutant renin gene in familial elevation of prorenin. J Biol Chem.1994; 269:30307–12.[ PubMed : 7982942]
- Wilcox WD. Abnormal serum uric acid levels in children. J Pediatr.1996; 128:731–41.[ PubMed : 8648529]
- Wolf MT, Hildebrandt F. Nephronophthisis. Pediatr Nephrol.2011; 26:181–94.[ PMC free article : PMC4160028] [ PubMed : 20652329]
- Zivná M, Hůlková H, Matignon M, Hodanová K, Vylet'al P, Kalbácová M, Baresová V, Sikora J, Blazková H, Zivný J, Ivánek R, Stránecký V, Sovová J, Claes K, Lerut E, Fryns JP, Hart PS, Hart TC, Adams JN, Pawtowski A, Clemessy M, Gasc JM, Gübler MC, Antignac C, Elleder M, Kapp K, Grimbert P, Bleyer AJ, Kmoch S. Dominant renin gene mutations associated with early-onset hyperuricemia, anemia, and chronic kidney failure. Am J Hum Genet.2009; 85:204–13.[ PMC free article : PMC2725269] [ PubMed : 19664745]
章节注释
作者介绍
Stanislav Kmoch博士 ( mailto:dev@null) 和Anthony Bleyer博士 ( mailto:dev@null) 积极参与有关renin基因突变和其它形式的遗传性肾脏疾病患者的临床研究,并十分乐意与对诊断或其它方面有疑问的患者进行交流。
修订沿革
- 2015/12/29 (me) 全面更新实时发布
- 2011/4/5 (me) 综述实时发布
- 2010/12/21 (ab) 首稿