
概述
临床特征
泛酸激酶相关神经变性 (PKAN) 是一种伴有脑铁积累 (NBIA) 的神经变性。 PKAN 的表型谱包括经典 PKAN 和非典型 PKAN。经典 PKAN 的特点是儿童早期出现进行性肌张力障碍、构音障碍、强直和舞蹈手足徐动症。色素性视网膜变性很常见。非典型 PKAN 的特点是发病较晚(年龄 > 10 岁)、明显的言语缺陷、精神障碍和更渐进的疾病进展。
诊断/测试
PKAN 的诊断基于先证者具有特征性临床特征和脑 MRI 上识别的“老虎眼”征( 冠状面T2 加权图像苍白球部位铁沉积显示低信号,而在苍白球的前内侧显示高信号)。分子遗传学检测的鉴定双等位基因的PANK2 致病性变异证实诊断。
管理
对症治疗:肌肉注射肉毒杆菌毒素、消融性苍白球切开术或丘脑切开术、鞘内注射或口服巴氯芬、口服苯海索、深部脑刺激、物理治疗和职业治疗以保持关节活动度、为步态异常转诊适应性辅助设备(助行器、轮椅)、言语治疗和/或辅助通讯设备、眼科治疗视网膜病变、转介社区资源:金融服务、盲人服务和教育计划。
预防继发并发症:严重口颊舌肌张力障碍导致反复咬舌时进行全口拔牙;根据需要进行胃造口管喂养。
监测:评估极度痛苦发作期间可治疗的疼痛原因;监测身高和体重;常规眼科评估;创伤的口头评估、步行和语言能力评估、喂养和营养评估。
遗传咨询
PKAN 以常染色体隐性遗传方式遗传。在受孕时,受累的个体的每个同胞有 25% 的风险患病,有 50% 的机会成为无症状携带者,以及 25% 的机会不患病也不是携带者。如果在受影响的家庭成员中发现了两种致病变异,则可以对有风险的亲属进行携带者检测和对有风险的妊娠进行产前检测。
GeneReview 谱
| 泛酸激酶相关神经变性:包括的表型 1 |
|---|
|
有关同义词和过时的名称,请参见 Nomenclature.
- 1.
对于这些表型的其他遗传原因,请参见 Differential Diagnosis.
诊断
提示性发现
具有以下临床、影像学和实验室特征以及家族史的个体应怀疑泛酸激酶相关神经变性 (PKAN)。
临床表现- 肌张力障碍
- 构音障碍
- 痉挛
- 舞蹈手足徐动症
- 帕金森症
- 反射亢进
- 伸脚趾迹象
- 发病于生命的第一个到第三个十年
- 步态改变/不能行走
- 色素性视网膜病变
- 智力和发育障碍,主要发生在小龄的儿童中
影像学特征 T2 加权脑 MRI 上的“虎眼”征(≥1.5Tesla):苍白球冠状或横向 T2 加权图像上的高信号中心区域被低信号边缘包围。 请参见 Figure 1.
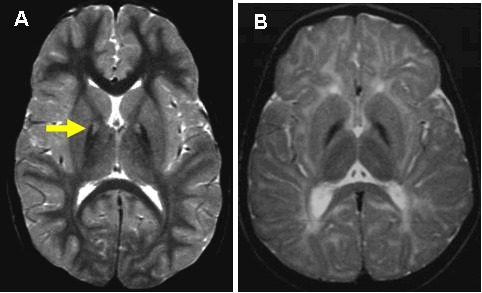
Figure 1.
PKAN(A)和非PKAN NBIA(B)的T2加权脑MRI A.箭头表示PKAN的“虎眼”变化特征。
实验室特点
- 棘红细胞增多症。据报道,一部分 PKAN 患者中存在棘细胞 [Schiessl-Weyer et al 2015]。对棘细胞进行严格的分析在技术上是困难的,并且由于分子遗传学诊断已经使用了几年,因此很少使用。
- 血浆前β脂蛋白部分低或缺失。见Clinical Description.
排除性发现
- 血浆铜蓝蛋白浓度或铜代谢异常(见 Wilson Disease)
- 通过电子显微镜、酶测定的神经元蜡样脂褐质沉积症的证据或与该病症相关的任何基因中存在致病性变异
- Beta-hexosaminidase A deficiency (β-己糖胺酶 A 缺乏症)或 GM1-半乳糖苷酶缺乏症
- 周围神经系统中球状体的病理学证据,表明PLA2G6-associated neurodegeneration (PLAN) 或mitochondrial membrane protein-associated neurodegeneration (MPAN)
建立诊断
PKAN 的诊断建立在具有以下标志性特征的先证者中。诊断标准不断发展,以反映 PKAN 与其他形式的neurodegeneration with brain iron accumulation (NBIA) 之间的区别。如果临床特征尚无定论,则通过 分子遗传学检测鉴定 PANK2 中 双等位基因的致病性变异可确认诊断(见Table 1)。
经典和非典型 PKAN 的标志性特征(见Figure 1)
- 锥体外系功能障碍,包括以下一项或多项:
- 肌张力障碍
- 痉挛
- 舞蹈手足徐动症
- 发作
- 经典形式. 通常在生命的头十年
- 非典型形式. 更常见于生命的第二个或第三个十年
- 丧失行走能力
- 经典形式. 通常在发病后 10 到 15 年内发生
- 非典型形式. 可能在发病后 15 至 40 年内发生
- T2 加权脑 MRI 上的“虎眼”征(≥1.5 Tesla):苍白球冠状或横向 T2 加权图像上的高信号中心区域被低信号边缘包围 (Figure 1)
注意:(1) 在疾病的早期阶段可能不存在该体征[Chiapparini et al 2011]. (2) 高信号区域被铁取代,随着时间的推移变得更加均匀低信号 [Delgado et al 2012]. (3) 多米尼加共和国的一些受累的者缺乏“虎眼标志” [Delgado et al 2012]. (4) 一些据称有虎眼征的人被诊断为 MPAN.
分子遗传学检测方法可以包括单基因检测、使用multigene panel和全基因组的检测:
multigene panel 包含 PANK2 和其他可能的基因(见Differential Diagnosis)的多基因套餐。注:(1)panel 中包含的基因和用于每个基因的检测的诊断工具敏感性因实验室而异,并且可能随时间而变化。 (2) 一些多基因 panel 可能包含与本 GeneReview 中讨论的疾病无关的基因;因此,临床医生需要确定哪种多基因最有可能以最合理的成本识别疾病的遗传原因,同时限制不能解释 表型和意义不确定变异的出现。 (3) 在一些实验室中,panel 选项可能包括定制的实验室设计的panel 和/或定制的以表型为中心的外显子组分析,其中包括临床医生指定的基因。 (4) panel 中使用的方法可能包括序列分析, deletion/duplication analysis和/或其他非基于序列的测试。
有关多基因套餐的介绍,请单击此处。有关订购基因检测的临床医生的更多详细信息,请参见此处。
Table 1
PKAN 和 NBIA 中使用的分子遗传学检测
| 基因 1 | 方法 | 测试方法可检测到的致病变异的比例 2 |
|---|---|---|
| PANK2 | 测序 3 | >99% of 患有 NBIA 的人在 MRI 上出现“虎眼”迹象4, 5 ~50% of 临床诊断为 NBIA 的个体 |
| 基因靶向 deletion/duplication analysis 6 | ~3%-5% 7 |
- 1.
染色体 位点和蛋白质信息见 Table A. Genes and Database
- 2.
有关在该基因中检测到的等位基因变异的信息见 Molecular Genetics
- 3.
- 4.
NBIA 国际突变数据库,俄勒冈健康与科学大学,未发表的数据
- 5.
- 6.
基因靶向 deletion/duplication analysis检测基因内缺失或重复。使用的方法可能包括 quantitative PCR、长程 PCR、多重连接依赖性探针扩增 (MLPA) 和设计用于检测单外显子缺失或重复的基因靶向微阵列。
- 7.
临床特征
临床表现
经典泛酸激酶相关神经变性 (PKAN)
经典 PKAN 的临床特征非常一致。它出现在儿童早期,通常在六岁之前(平均年龄:3.4 岁)。最常见的症状是下肢肌张力障碍和痉挛共同导致的步态障碍,以及视网膜病变儿童的视野受限。有些孩子有发育迟缓,主要是运动性的,但有时是全身性的。ADHD或踮脚尖走的早期病史也很常见。视觉症状可能会使患有 PKAN 的儿童就医。
神经 经典 PKAN 的神经系统体征和症状主要是锥体外系,包括肌张力障碍和构音障碍。肌张力障碍总是存在并且通常是早期表现。颅肌张力障碍和肢体肌张力障碍很常见,并且可能分别导致反复的舌头创伤和由极端骨应力和骨质减少共同导致的非创伤性长骨骨折。由此产生的疼痛和痛苦可能会在一个难以打破的循环中导致肌张力障碍的发展。
皮质脊髓束受累很常见,包括痉挛、反射亢进和伸趾征。
癫痫发作很少见。
智力障碍是 PKAN 的一个特征,尤其是在发病年龄小的儿童中。一项针对 16 名患有 PKAN 的儿童和成人的研究显示,通过标准化评估工具测量的认知表达不同,技能从高平均水平到明显低于平均水平不等。发病年龄与智力障碍有很强的负相关(即,早期发病与更大的障碍相关)[Freeman et al 2007]. 然而,患有典型疾病的儿童保留了已获得的认知能力,并且不会随着后来的运动功能丧失而失去这些技能。
视网膜变性。三分之二的受累的经典 PKAN 患者会出现色素性视网膜变性。视网膜病变发生在疾病的早期,尽管在进行包括视网膜电图 (ERG) 和视野测试在内的全面诊断评估之前通常不会发现它。视网膜变性遵循典型的临床过程,伴有夜盲症(夜盲症),随后周边视野逐渐丧失,有时最终失明。眼底变化最初包括有斑点的视网膜,后来发展为骨针形成、明显的脉络膜脉管系统和“牛眼”环形黄斑病变。在诊断时眼科检查正常的个体通常不会在以后发生视网膜病变。
异常眼球运动,包括垂直扫视和扫视追踪,很常见。在一项研究中,10 名 PKAN 患者中有 8 名患有扇形性虹膜麻痹和瞳孔环部分缺失,与双侧Adie's 瞳孔一致[Egan et al 2005].在 PKAN 中很少见到视神经萎缩。
预后。 PKAN 是一种进行性疾病。失去的技能通常不会重新获得。进展速度与发病年龄相关:那些有早期症状的人下降得更快。随着疾病的进展,肌张力障碍和痉挛会影响孩子的行走能力;大多数患有早发性疾病的人都是十几岁左右坐轮椅,有些人更早。 PKAN 以不均匀的速度发展。受影响的个体经历快速恶化的发作,通常持续一到两个月,穿插更长的稳定期。压力和代谢的常见原因似乎与衰退期无关,这种现象尚未找到原因。
确实会发生过早死亡。然而,随着医疗保健的改善,越来越多的 受累的患者活到成年。口面部肌张力障碍可导致吞咽困难和营养不良的继发影响。与原发性神经退行性过程相比,过早死亡更可能与这些继发性影响(例如营养相关免疫缺陷、吸入性肺炎)有关。在极少数情况下,死亡发生在肌张力障碍状态。
非典型 PKAN
非典型 PKAN 的临床特征比经典 PKAN 更多样化。发病时间为前 30 年(平均年龄:13.6 岁)。非典型形式的进展较慢,呈现特征明显,通常涉及言语作为唯一呈现特征或表现之一。言语缺陷包括 palilalia(单词或短语的重复)、tachylalia/tachylogia(快速单词和/或短语)和构音障碍(发音不佳、口齿不清)。
在非典型 PKAN 中常见的精神症状包括性格改变、冲动和暴力爆发、抑郁和情绪不稳定。受影响的个体还可能表现出运动和语言抽搐、强迫行为,以及罕见的精神病症状[del Valle-López et al 2011].
与经典 PKAN 一样,非典型 PKAN 患者可能会出现认知障碍,但需要进行额外调查。Freeman et al [2007] 发现,较晚的发病年龄与较少的智力和适应性行为障碍相关。
运动受累通常是后来的特征,尽管运动受累的个体在儿童期和青春期通常被描述为笨拙。痉挛、反射亢进和其他皮质脊髓束受累的迹象很常见,最终限制了行走。明显地让人想起Parkinson disease,观察到行走过程中的“冻结”(尤其是在转弯或遇到表面变化时)[Guimarães & Santos 1999].
还报道了一种特发性震颤样综合征[Yamashita et al 2004].
视网膜病变在非典型 PKAN 中很少见,并且视神经萎缩与非典型 PKAN 无关。
HARP 综合征(低前β脂蛋白血症、棘细胞增多症、色素性视网膜炎和苍白球变性)。最初被描述为具有单独的临床表现的疾病,有报道HARP 综合征的两个家族现在已知为PKAN 的表型谱。
基因型-表型相关性
然而,具有两种无效 变异(预测不产生蛋白质)的个体始终具有经典的 PKAN。致病变异的其他组合(即无效/错义, 纯合性错义或复合杂合错义)以不可预测的模式产生经典或非典型表型。
致病性 错义变异p.Gly521Arg的纯合性始终表现为经典的 PKAN;然而,与其他常见等位基因纯合性相关的 表型是不可预测的。三分之二的 PKAN 患者是复合杂合子,临床病程不可预测。
在家庭中, 受累的个体之间的表型相当一致。在患有非典型 PKAN 的家庭中,发病年龄、表现特征和进展速度的差异更大。
命名法
鉴于两位德国神经病理学家在二战之前和期间的不道德活动,同名的 Hallervorden-Spatz 综合征 (HSS) 不再受到青睐 [Shevell 2003].
患病率
没有收集到关于这种罕见疾病的可靠患病率数据。有人建议估计 1,000,000 人中有 1 到 3 人。这个数字意味着一般人群携带者的频率为 1:275-1:500。
在荷兰描述了一种建立者效应[Rump et al 2005]。多米尼加共和国西南部的一个社区也有一个共同的建立者变异: c.680A>G (p.Tyr227Cys)。在这个小而孤立的人群中,携带者的频率明显增加[Delgado et al 2012].
遗传相关(等位基因)疾病
已知没有其他表型与 PANK2 中的致病变异相关。
鉴别诊断
泛酸激酶相关神经变性 (PKAN) 是一种 neurodegeneration with brain iron accumulation(NBIA). NBIA 被定义为一组进行性锥体外系疾病,影像学证据表明大脑中局灶性铁积累,通常在基底节。 NBIA 的类型包括在Table 2中。
脑铁积累神经变性多基因套餐可能包括测试与本节讨论的疾病相关的许多基因。 注意:多基因 panel 中包含的基因和使用的方法因实验室而异,并且可能会随着时间而改变;套餐可能不包含特定的可能基因。
Table 2
NBIA 的类型:分子遗传学
| 基因 | 疾病名称 | MOI | 进程 |
|---|---|---|---|
| ATP13A2 | Kufor-Rakeb 综合征 1 | AR | 迟发性、缓慢进展的 NBIA,在第 1 个十年后发病 |
| C19orf12 | MPAN | AR | 早发(第 1 个十年期间)或晚发(第 1 个十年后),缓慢进展的 NBIA |
| CP | Aceruloplasminemia | AR | 迟发性、缓慢进展的 NBIA,在第 1 个十年后发病 |
| CoASY | CoPAN | AR | 早发、缓慢进展的 NBIA,在第 1 个十年内发病 |
| DCAF17 | Woodhouse-Sakati syndrome | AR | 早发、缓慢进展的 NBIA,在第 1 个十年内发病 |
| FA2H | FAHN | AR | 早发、缓慢进展的 NBIA,在第 1 个十年内发病 |
| FTL | Neuroferritinopathy | AD | 迟发性、缓慢进展的 NBIA,在第 1 个十年后发病 |
| PANK2 | Atypical PKAN | AR | 迟发性、缓慢进展的 NBIA,在第 1 个十年后发病 |
| Classic PKAN | 早发、缓慢进展的 NBIA,在第 1 个十年内发病 | ||
| PLA2G6 | Atypical neuroaxonal dystrophy | AR | 早发、缓慢进展的 NBIA,在第 1 个十年内发病 |
| Infantile neuroaxonal dystrophy | 早发、快速进展的 NBIA,在第 1 个十年内发病 | ||
| PLA2G6-associated dystonia-parkinsonism | 迟发性、缓慢进展的 NBIA,在第 1 个十年后发病 | ||
| WDR45 | BPAN | XL | 早发、快速进展的 NBIA,在第 1 个十年内发病 |
| 不详 | 原发NBIA | 迟发性、缓慢进展的 NBIA,在第 1 个十年后发病 |
- 1.
一些 Kufor-Rakeb 综合征患者的脑铁含量较高 [Schneider et al 2010].
PKAN 可以通过以下发现与其他形式的 NBIA 区分开来:
- 脑部核磁共振
- 在大多数非 PKAN NBIA 患者中,苍白球在 T2 加权图像上呈均匀低信号(见 Figure 1),表明铁含量高。这种变化与“虎眼”标志不同,并且与 PANK2 中的致病变异无关。应该注意的是,在 MPAN 中,内侧和外侧之间的内侧髓板的高信号条纹可能类似于“虎眼”征 [Hogarth et al 2013].
- 红核和齿状核中的铁沉积见于神经铁蛋白病和无铜蓝蛋白血症。小脑萎缩在 PLAN 中很常见。
- PKAN 和 BPAN 均报告了通过 CT 扫描检测到的双侧苍白球钙化 [Wu et al 2013, Fasano et al 2017].
- PKAN 中没有癫痫发作;在某些形式的非 PKAN NBIA 中癫痫发作突出
经典的 PKAN。四种疾病可能表现出与经典 PKAN 相似的早期临床变化:
- 利氏综合症。 T2 加权 MRI 上苍白球中的对称高信号可能类似于“老虎眼”征,但缺乏由铁积累引起的周围低信号。与 PKAN 不同,对称性高信号经常发生在基底节的其他区域(参见Nuclear Gene-Encoded Leigh Syndrome Overview and Mitochondrial DNA-Associated Leigh Syndrome and NARP)。
- 婴儿神经轴索营养不良(Infantile neuroaxonal dystrophy,INAD). 部分个体在苍白球和黑质中表现出低信号,但“虎眼”征不存在,小脑萎缩很常见。在 INAD 中,轴突球体存在于周围神经系统中,而在 PKAN 中,它们仅位于中枢神经系统中。
非典型 PKAN。鉴别诊断包括以下内容:
- 早发性帕金森病,包括 parkin type of juvenile Parkinson disease和 PLA2G6 相关肌张力障碍-帕金森病,最初可能与非典型 PKAN 相似,发病年龄在 20 至 40 岁之间,并伴有下肢肌张力障碍。运动迟缓和静止性震颤也是常见特征。
- 原发性家族性脑钙化(Primary familial brain calcification,PFBC). 受影响的个体在基底神经节中有异常的钙沉积,包括在苍白球中可能类似于“老虎眼”标志的沉积。 PKAN 的共同特征包括帕金森症、构音障碍、肌张力障碍和痉挛。钙沉积物随着时间的推移在基底神经节和大脑皮层中积累,有助于将其与 PKAN 区分开来。据报道,PDGFB、PDGFRB 和 SLC20A2 的致病性变异会导致 PFBC;遗传是常染色体显性遗传.
- 神经铁蛋白病(Neuroferritinopathy)通常在 4 到 50 岁时出现不自主运动,并且在 PKAN 中没有表现出明显的构音障碍。
- 原发性精神疾病。在没有构音障碍的情况下存在冲动和其他行为变化可能表明存在原发性精神疾病。对于此类中的所有疾病,T2 加权脑 MRI 将根据“老虎眼”标志的存在来区分 PKAN。
其他需要考虑的疾病:
- 神经元蜡样脂褐质沉着症
- 儿童期遗传性共济失调( ataxias)(尤其是 SCA3和SCA7)
- DYT1等肌张力障碍
- 少年Huntington disease
- 舞蹈病-棘红细胞增多症(Chorea-acanthocytosis)
- Lesch-Nyhan 综合征(Lesch-Nyhan syndrome)
- 威尔逊病(Wilson disease)
- 隐性遗传性痉挛性截瘫(Recessive hereditary spastic paraplegia)
- 抽动秽语障碍 [Scarano et al 2002]
神经棘红细胞增多症综合征。与红细胞棘红细胞增多症相关的神经系统疾病称为神经棘红细胞增多症综合征。
一组神经棘红细胞增多症与脂质吸收不良有关,主要影响脊髓、小脑和周围神经系统。神经系统发现包括以下内容:
- 一种渐进性脊髓小脑退化,伴步态共济失调、视力障碍和构音障碍
- 一种脱髓鞘感觉运动和轴突周围神经病,伴有反射减退、振动和位置觉减弱
- 锥体束征(罕见)
- 颅神经受累(罕见)
这些疾病包括:
- 1 型低β脂蛋白血症 (FHBL1; OMIM 615558)
- 2型低β脂蛋白血症 (FHBL2; OMIM 605019)
- 无β脂蛋白血症(Abetalipoproteinemia,ABL,Bassen-Kornzweig 病)
FHBL1、FHBL2 和 ABL 在棘红细胞增多症、构音障碍、神经病变和反射消失方面具有相同的发现,但不同之处在于 ABL、FHBL1 和 FHBL2 有色素性视网膜病变并且没有基底节受累。 ABL、FHBL1 和 FHBL2 是由影响微粒体甘油三酯转移蛋白的致病变异引起的,导致维生素 E 缺乏。 ABL 以 常染色体隐性遗传方式遗传。 FHBL1和FHBL2在纯合性和杂合的状态下均有临床表现。
第二组神经棘红细胞增多症综合征主要影响中枢神经系统,特别是基底神经节,导致类似于Huntington disease的舞蹈症候群。这些疾病包括:
- 麦克劳德神经棘红细胞增多症 (McLeod neuroacanthocytosis syndrome,MLS) 是一种具有血液、神经肌肉和中枢神经系统 (CNS) 表现的多系统疾病。受影响的男性有 McLeod 血型表型和红细胞棘红细胞增多症。 MLS 的神经肌肉表现包括亚临床或轻度感觉运动轴索病、肌病和心肌病。 MLS 的 CNS 表现类似于亨廷顿病,包括舞蹈样运动障碍、“皮质下”认知缺陷、精神病表现,以及在某些个体中,癫痫发作。 XK 是已知致病性变异导致 MLS 的唯一 基因;遗传方式是 X-linked。
- 舞蹈症-棘红细胞增多症(Chorea-acanthocytosis,ChAc) 特征是舞蹈症、肌病、进行性认知和行为改变以及癫痫发作。平均发病年龄约为 35 岁,尽管 ChAc 可早于第一个十年或迟至第七个十年发展。 VPS13A 是目前已知的唯一突变导致 ChAc 的基;遗传是常染色体隐性遗传.
- 亨廷顿病样 2(Huntington disease-like 2,HDL2)在 3 至 40 岁出现,并在 10 至 15 年内呈渐进过程。肌张力障碍是常见的发现;舞蹈病或帕金森症可能会随着疾病的发展而改变。迄今为止,几乎所有 受累的人都有非洲血统。红细胞棘红细胞增多症是可变的。 JPH3 是唯一已知突变会导致 HDL2 的基因;遗传是 常染色体显性遗传.
处理
初步诊断后的评估
为了确定被诊断患有泛酸激酶相关神经变性 (PKAN) 的个体的疾病程度,如果尚未完成,建议执行以下操作:
- 肌张力障碍、强直、舞蹈手足徐动症和痉挛的神经系统检查,包括对行走和言语的评估
- 眼科评估视网膜病变的证据
- 筛选发育评估,如果表明延迟,则转诊进行更正式的测试
- 物理治疗、职业治疗和/或言语治疗的评估
- 咨询临床遗传学家和/或遗传咨询师
治疗表现
PKAN 的共识临床管理指南可用于提供详细的管理信息 [Hogarth et al 2017].药理学和外科干预的重点是缓解症状。
对症治疗主要针对肌张力障碍,这可能会使 受累的个人和护理人员极度疲劳和痛苦。用于控制受影响个体的肌张力障碍的疗法取得了不同程度的成功,包括:
- 肌肉注射肉毒杆菌毒素
- 口服巴氯芬、苯海索和氯硝西泮:对 PKAN 最有效的一线药物
- 二线药物包括可乐定、加巴喷丁、丁苯那嗪和普瑞巴林
- 鞘内和脑室内巴氯芬
- 深部脑刺激,在临床上使用的频率越来越高,并有一些初步获益的证据,尽管它可能不会随着疾病的进展而持续 [Lim et al 2012, Garcia-Ruiz et al 2015, Hogarth et al 2017]
- 消融性苍白球切开术或丘脑切开术。这些消融程序主要被 DBS 取代,但在某些个体中可能仍然有用 [Dwarakanath et al 2014].
- 张力障碍状态(张力障碍风暴)的紧急医疗(通常住院),这是一种常见的现象。 PKAN 共识指南提供了有关张力障碍风暴的方法和管理的详细信息 [Hogarth et al 2017].
- 指定的物理和职业治疗,特别是对于那些只有轻微症状的人。尽可能长时间保持正常关节活动度的治疗可能是有用的。
- 根据需要转诊适应性辅助设备(例如,步行者或轮椅因步态异常)
- 用于 PKAN 相关构音障碍和语言延迟的语言治疗和/或辅助通信设备
其他表现
- 根据眼科治疗和干预视网膜病变
- 转介到适当的社区资源以获得金融服务、盲人服务(如果存在视网膜病变)和特殊教育
预防继发性并发症
严重口颊舌肌张力障碍引起的反复咬舌是 PKAN 中难以应对的特殊挑战。可以制作定制的咬锁矫正器具并将其粘合到位,以防止舌头撕裂伤。应尽一切努力避免完全拔牙。
一旦个体由于吞咽困难或呼吸系统并发症而不能再通过口服维持足够的饮食,就需要放置胃造口管。
在典型疾病的晚期阶段,也可能需要气管切开术。
监测
随着疾病的进展,极度痛苦的发作可能会持续数天或数周。在这些发作期间评估可治疗的疼痛原因尤为重要。这些可能包括隐匿性胃肠道出血、尿路感染、口腔撕裂伤和隐匿性骨折。非行走个体的骨质减少与肌张力障碍对长骨的明显压力相结合,使患有 PKAN 的个体在没有明显外伤的情况下发生骨折的风险特别高。
应定期执行以下操作:
- 使用适当的生长曲线监测身高和体重以筛查儿童的营养状况恶化
- 眼科评估
- 创伤后果的口头评估
- 评估步行、环境适应、言语能力和沟通需要,以帮助受累的人保持独立
- 吞咽评估和定期饮食评估以确保充足的营养
要避免的药物/情况
三名患有非典型 PKAN 的同胞接受 α-生育酚和艾地苯醌治疗后的报告表明症状恶化,一旦停止使用这些化合物,症状就会有所改善 [JP Harpey,个人通讯]。
风险亲属的评估
请参阅Related Genetic Counseling Issues.
正在研究的疗法
铁螯合。随着去铁酮 (Ferriprox®) 在几个受累的个体群体中的数据积累,对铁螯合的兴趣重新出现。与早期药物不同,去铁酮可穿过血脑屏障并去除细胞内铁。已经进行了一项小型 II 期试点试验,以评估 PKAN 人群中的去铁酮。完成研究的九名受影响个体对去铁酮的耐受性良好,通过 MRI 评估,苍白球中的铁含量在统计学上明显减少。然而,他们的临床状况没有变化。作者建议可能需要更长的试验期才能产生临床改善 [Zorzi et al 2011].最近完成了一项关于去铁酮的国际随机、双盲、安慰剂对照试验,目前正在分析数据 (clinicaltrials.gov).
附加疗法。目前正在为 PKAN 开发多种化合物,预计将进入临床试验。临床医生应定期查看美国的ClinicalTrials.gov和欧洲的 EU Clinical Trials Register,并与 PKAN 研究人员保持联系。
其他
泛酸。一些 PKAN 患者存在残留酶活性,这增加了使用高剂量泛酸(PANK2 酶底物)进行治疗的可能性。泛酸盐对人体没有已知的毒性;口服高剂量的泛酸或泛酸钙(≤10 克/天,持续数周)似乎对人体无毒。泛酸补充剂在改善症状方面的功效目前尚不清楚;一些患有非典型病程的人据传在服用泛酸后症状(构音障碍、步态不平衡、幸福感)有所改善。
二十二碳六烯酸 (DHA)。基于辅酶 A 在脂肪酸合成和降解中的作用,DHA 作为视杆细胞视盘膜主要成分的重要性,以及对大部分 PKAN 患者视网膜变性的观察,DHA 可能具有一定的作用在预防这种并发症方面,尽管尚未进行任何研究。该化合物可以 omega-3 脂肪(鱼油)的形式作为口服营养补充剂提供,并且没有已知的毒性。
其他治疗
- 可能对其他形式的 NBIA 起作用但通常对 PKAN 患者无帮助的疗法包括左旋多巴/卡比多巴和溴隐亭。
- 用泛酸激酶的产物磷酸泛酸治疗 PKAN ,由于缺乏可用的化合物以及关于其对人类或动物的安全性或毒性的任何信息而变得复杂。此外,磷酸泛酸不太可能很容易地跨细胞膜转运,这使得这种假设治疗的成功值得怀疑。
遗传咨询
遗传咨询是向个人和家庭提供有关遗传疾病的性质、遗传和影响的信息,以帮助他们做出明智的医疗和个人决定的过程。 以下部分涉及遗传风险评估以及使用家族史和基因检测来阐明家庭成员的遗传状况。 本节并不旨在解决个人可能面临的所有个人、文化或伦理问题,也不能替代遗传学专家的咨询。 -编者。
遗传方式
泛酸激酶相关神经变性 (PKAN) 以常染色体隐性遗传方式遗传。
家庭成员的风险
先证者的父母
先证者的兄弟姐妹
先证者的后代
其他家庭成员。先证者父母的每个同胞有 50% 的风险成为携带者.
携带者(杂合子)检测
对高危亲属进行携带者检测需要事先鉴定家族中的 PANK2 致病变异。
相关遗传咨询问题
检测无症状的高危同胞,尤其是那些比先证者年轻的同胞。对于先证者看似健康的同胞,尤其是当他们比先证者年轻时,可以考虑进行神经系统评估(包括脑部 MRI)和基因检测。尽管过去不鼓励对 18 岁以下的无症状个体进行评估和检测,但鉴于正在开发的治疗方法和正在进行的去铁酮临床试验的数据分析,可能更常考虑这样做。
家庭计划
DNA 银行是 DNA(通常从白细胞中提取)的存储,以备将来使用。 由于测试方法以及我们对基因、等位基因变异和疾病的理解在未来可能会得到改善,因此应考虑将受累的患者的 DNA 银行化。
产前检测和植入前遗传学诊断
资源
GeneReviews 工作人员选择了以下特定疾病和/或支持组织和/或登记处,以造福患有这种疾病的个人及其家人。 GeneReviews 不对其他组织提供的信息负责。 有关选择标准的信息,请单击 here.
- My46 Trait Profile
- National Library of Medicine Genetics Home Reference
- NBIA AllianceEmail: Info@NBIAalliance.org
- NBIA Disorders Association2082 Monaco CourtEl Cajon CA 92019-4235Phone: 619-588-2315Fax: 619-588-4093Email: info@NBIAdisorders.org
- eyeGENE - National Ophthalmic Disease Genotyping Network RegistryPhone: 301-435-3032Email: eyeGENEinfo@nei.nih.gov
- NBIA Disorders Association Research Registry and Treat Iron-Related Childhood-Onset Neurodegeneration (TIRCON) RegistryCA 92019-4235Phone: 619-588-2315Fax: 619-588-4093Email: pwood@nbiadisorders.org
- NBIAcureCenter of Excellence for NBIA Clinical Care and ResearchInternational Registry for NBIA and Related DisordersOregon Health & Science UniversityPhone: 503-494-4344Fax: 503-494-6886Email: gregorya@ohsu.edu
- Neuroacanthocytosis Database (Registry)Prof. Adrian DanekNeurologische KlinikPOB 701260GermanyPhone: 49 (89) 4400-76676Email: adrian.danek@med.uni-muenchen.de
分子遗传
Molecular Genetics 和 OMIM 表格中的信息可能与 GeneReview 中其他地方的不同:表格可能包含更新的信息。 -编者。
Table A
泛酸激酶相关的神经变性:基因和数据库
| 基因 | 染色体定位 | 蛋白质 | 数据库 | HGMD | ClinVar |
|---|---|---|---|---|---|
| PANK2 | 20p13 | Pantothenate kinase 2, mitochondrial | PANK2 database | PANK2 | PANK2 |
Table B
泛酸激酶相关神经变性的 OMIM 条目(View All in OMIM)
分子发病机制
泛酸激酶相关神经变性 (PKAN) 归因于泛酸激酶 2 的缺陷或完全缺失,泛酸激酶 2 由四种人类泛酸激酶基因之一的 PANK2 编码。泛酸激酶缺乏被认为会导致 N-泛酰半胱氨酸和泛硫乙胺的积累,这可能直接或通过作为铁螯合剂的自由基损伤引起细胞毒性 [Yang et al 2000, Yoon et al 2000].泛酸激酶 2 缺乏可能会导致辅酶 A (CoA) 耗竭和膜生物合成缺陷,在这些组织中辅酶 A (CoA) 是主要的泛酸激酶,或者在 CoA 需求量最大的组织中。
杆状感光器不断产生膜状圆盘;因此,在经典 PKAN 中经常观察到的视网膜病变可能继发于这种缺陷。导致临床后遗症的生化机理仍未完全了解,需要进一步研究。
基因结构。PANK2 编码一个 1.85-kb 的转录本,该转录本来自七个外显子,跨越超过 35 Mb 的基因组的DNA。详细的序列分析表明,PANK2 是真核基因家族的成员,该家族由一组六个外显子组成,这些外显子编码同源核心蛋白,前面是一系列替代起始外显子,其中一些外显子编码独特的氨基末端肽。替代剪接, 涉及使用替代的第一个外显子,导致多个转录本编码不同的异型体.有关 基因和蛋白质信息的详细摘要,请参见 Table A,基因。
致病变异。除了 Table 3中描述的三种常见的 PANK2 致病变异外,致病变异通常是每个家族所私有的并且类型各不相同。
Table 3
特定的PANK2 致病变异
| DNA 核酸改变 (Alias 1) | 蛋白质改变 (Alias 1) | 参考序列 2 |
|---|---|---|
| c.680A>G | p.Tyr227Cys | NM_153638 NP_705902 |
| c.1351C>T 3 (1021C>T) | p.Arg451Ter 3 (Arg341Ter) | |
| c.1561G>A (1231G>A) 3, 4 | p.Gly521Arg 3, 4 (Gly411Arg) | |
| c.1583C>T 3 (1253C>T) | p.Thr528Met 3 (Thr418Met) | |
| c.1413-1G>T 5 (IVS4-1G>T) | --- |
关于变异分类的注意事项:表中列出的变异由作者提供。 GeneReviews工作人员尚未独立验证变异的分类。
关于命名法的注释:GeneReviews遵循人类基因组变异学会(varnomen- .hgvs.org )的标准命名惯例。 有关术语的解释,请参见Quick Reference。- 1.
不符合当前命名约定的变异名称
- 2.
参考序列是最长的异构体,PANK2 异构体 1 前原蛋白。
- 3.
常见致病性变异 (等位基因频率):p.Gly521Arg (25%); p.Thr528Met (8%); p.Arg451Ter (3%)
- 4.
该等位基因的纯合性导致典型疾病。
- 5.
导致 PKAN 的致病性变异,最初见于被诊断患有 HARP 综合征的个体 [Ching et al 2002]
正常基因产物.PANK2 编码50.5-kd 蛋白质,它是一种功能性泛酸激酶[Zhou et al 2001]. 泛酸激酶是辅酶 A 生物合成中必不可少的调节酶,可催化泛酸(维生素 B5)、N-泛酰半胱氨酸和泛硫乙胺的磷酸化。泛酸激酶受原核生物中酰基辅酶 A 水平和真核生物中乙酰辅酶 A 水平的调节。
异常基因产物。致病变异通常可分为无效或错义等位基因。具有纯合性无效等位基因的个体通常患有典型疾病。目前尚不清楚具有非典型 PKAN 的个体是否具有部分酶功能。对于那些与致病性错义变异 复合杂合的人,假定存在等位基因间互补。当蛋白质亚基之间相互作用的结构域中的致病变异能够恢复部分功能时,就会产生等位基因间互补。这在理论上是特定于变异的,一些变异排除了互补。因此,一些错义变异的复合杂合子可能表现为典型疾病,而另一些则具有更不典型的病程。最近一项针对受累的个体的 PANK2 致病性变异的研究证实,最常见的 PANK2 致病性变异p.Gly521Arg 导致蛋白质错误折叠且缺乏活性 [Zhang et al 2006]. 然而,发现其他九种致病变异导致蛋白质具有正常的催化活性和调节功能。作者认为 PANK2 蛋白可能具有尚未被重视的其他功能。
参考文献
引用文献
- Chiapparini L, Savoiardo M, D’Arrigo S, Reale C, Zorzi G, Zibordi F, Cordelli DM, Franzoni E, Garavaglia B, Nardocci N. The "eye-of-the-tiger" sign may be absent in the early stages of classic pantothenate kinase associated neurodegeneration. Neuropediatrics. 2011;42:159 - 62. [PubMed: 21877312]
- Ching KH, Westaway SK, Gitschier J, Higgins JJ, Hayflick SJ. HARP syndrome is allelic with pantothenate kinase-associated neurodegeneration. Neurology. 2002;58:1673 - 4. [PubMed: 12058097]
- Delgado RF, Sanchez PR, Speckter H, Then EP, Jiminez R, Oviedo J, Dellani PR, Foerster B, Stoeter P. Missense PANK2 mutation without "eye of the tiger" sign: MR findings in a large group of patients with pantothenate kinase-associated neurodegeneration (PKAN). J Magn Reson Imaging. 2012;35:788 - 94. [PubMed: 22127788]
- del Valle-López P, Pérez-García R, Sanguino-Andrés R, González-Pablos E. Adult onset Hallervorden-Spatz disease with psychotic symptoms. Actas Esp Psiquiatr. 2011;39:260 - 2. [PubMed: 21769749]
- Dwarakanath S, Zafar A, Yadav R, Arivazhagan A, Netravathi M, Sampath S, Pal PK. Does lesioning surgery have a role in the management of multietiological tremor in the era of deep brain stimulation? Clin Neurol Neurosurg. 2014;125:131 - 6. [PubMed: 25128653]
- Egan RA, Weleber RG, Hogarth P, Gregory A, Coryell J, Westaway SK, Gitschier J, Das S, Hayflick SJ. Neuro-ophthalmologic and electroretinographic findings in pantothenate kinase-associated neurodegeneration (formerly Hallervorden-Spatz syndrome). Am J Ophthalmol. 2005;140:267 - 74. [PMC free article: PMC2169522] [PubMed: 16023068]
- Fasano A, Shahidi G, Lang AE, Rohani M. Basal ganglia calcification in case of PKAN. Parkinsonism Relat Disord. 2017;36:98 - 99. [PubMed: 28024710]
- Freeman K, Gregory A, Turner A, Blasco P, Hogarth P, Hayflick S. Intellectual and adaptive behavior functioning in pantothenate kinase-associated neurodegeneration. J Intellect Disabil Res. 2007;51:417 - 26. [PMC free article: PMC2099459] [PubMed: 17493025]
- Garcia-Ruiz PJ, Ayerbe J, Desojo L, Feliz C, Fernandez J. Deep brain stimulation for pantothenate kinase-associated neurodegeneration. Case Rep Neurol Med. 2015;2015:245735. [PMC free article: PMC4352941] [PubMed: 25802776]
- Guimarães J, Santos JV. Generalized freezing in Hallervorden-Spatz syndrome: case report. Eur J Neurol. 1999;6:509 - 13. [PubMed: 10362909]
- Hogarth P, Gregory A, Kruer MC, Sanford L, Wagoner W, Natowicz MR, Egel RT, Subramony SH, Goldman JG, Berry-Kravis E, Foulds NC, Hammans SR, Desguerre I, Rodriguez D, Wilson C, Diedrich A, Green S, Tran H, Reese L, Woltjer RL, Hayflick SJ. New form of neurodegeneration with brain iron accumulation: features associated with MPAN. Neurology. 2013;80:268 - 75. [PMC free article: PMC3589182] [PubMed: 23269600]
- Hogarth P, Kurian MA, Gregory A, Csányi B, Zagustin T, Kmiec T, Wood P, Klucken A, Scalise N, Sofia F, Klopstock T, Zorzi G, Nardocci N, Hayflick SJ. Consensus clinical management guideline for pantothenate kinase-associated neurodegeneration (PKAN). Mol Genet Metab. 2017;120:278 - 87. [PubMed: 28034613]
- Kruer MC, Hiken M, Gregory A, Malandrini A, Clark D, Hogarth P, Grafe M, Hayflick SJ, Woltjer RL. Novel histopathologic findings in molecularly-confirmed pantothenate kinase-associated neurodegeneration. Brain. 2011;134:947 - 58. [PMC free article: PMC3105492] [PubMed: 21459825]
- Lim BC, Ki CS, Cho A, Hwang H, Kim KJ, Hwang YS, Kim YE, Yun JY, Jeon BS, Lim YH, Paek SH, Chae JH. Pantothenate kinase-associated neurodegeneration in Korea: recurrent R440P mutation in PANK2 and outcome of deep brain stimulation. Eur J Neurol. 2012;19:556 - 61. [PubMed: 22103354]
- Rump P, Lemmink HH, Verschuuren-Bemelmans CC, Grootscholten PM, Fock JM, Hayflick SJ, Westaway SK, Vos YJ, van Essen AJ. A novel 3-bp deletion in the PANK2 gene of Dutch patients with pantothenate kinase-associated neurodegeneration: evidence for a founder effect. Neurogenetics. 2005;6:201 - 7. [PMC free article: PMC2105745] [PubMed: 16240131]
- Scarano V, Pellecchia MT, Filla A, Barone P. Hallervorden-Spatz syndrome resembling a typical Tourette syndrome. Mov Disord. 2002;17:618 - 20. [PubMed: 12112223]
- Schiessl-Weyer J, Roa P, Laccone F, Kluge B, Tichy A, De Almeida Ribeiro E, Prohaska R, Stoeter P, Salzer U. Acanthocytosis and the c.680 A>G mutation in the PANK2 gene: a study enrolling a cohort of PKAN patients from the Dominican Republic. PLos One. 2015;10:e0125861. [PMC free article: PMC4411072] [PubMed: 25915509]
- Schneider SA, Paisan-Ruiz C, Quinn NP, Lees AJ, Houlden H, Hardy J, Bhatia KP. ATP13A2 mutations (PARK9) cause neurodegeneration with brain iron accumulation. Mov Disord. 2010;25:979 - 84. [PubMed: 20310007]
- Shevell M. Hallervorden and history. N Engl J Med. 2003;348:3 - 4. [PubMed: 12510036]
- Swaiman KF. Hallervorden-Spatz syndrome. Pediatr Neurol. 2001;25:102 - 8. [PubMed: 11551740]
- Woltjer RL, Reese LC, Richardson BE, Tran H, Green S, Pham T, Chalupsky M, Light T, Sanford L, Jeong SY, Hamada J, Schwanemann LK, Rogers C, Gregory A, Hogarth P, Hayflick SJ. Pallidal neuronal apolipoprotein E in pantothenate kinase-associated neurodegeneration recapitulates ischemic injury to the globus pallidus. Mol Genet Metab. 2015;116:289 - 97. [PMC free article: PMC4688119] [PubMed: 26547561]
- Wu YW, Hess CP, Singhal NS, Groden C, Toro C. Idiopathic basal ganglia calcifications: an atypical presentation of PKAN. Pediatr Neurol. 2013;49:351 - 4. [PubMed: 23968566]
- Yamashita S, Maeda Y, Ohmori H, Uchida Y, Hirano T, Yonemura K, Uyama E, Uchino M. Pantothenate kinase-associated neurodegeneration initially presenting as postural tremor alone in a Japanese family with homozygous N245S substitutions in the pantothenate kinase gene. J Neurol Sci. 2004;225:129 - 33. [PubMed: 15465096]
- Yang EY, Campbell A, Bondy SC. Configuration of thiols dictates their ability to promote iron-induced reactive oxygen species generation. Redox Rep. 2000;5:371 - 5. [PubMed: 11140748]
- Yoon SJ, Koh YH, Floyd RA, Park JW. Copper, zinc superoxide dismutase enhances DNA damage and mutagenicity induced by cysteine/iron. Mutat Res. 2000;448:97 - 104. [PubMed: 10751627]
- Zhang YM, Rock CO, Jackowski S. Biochemical properties of human pantothenate kinase 2 isoforms and mutations linked to pantothenate kinase-associated neurodegeneration. J Biol Chem. 2006;281:107 - 14. [PubMed: 16272150]
- Zhou B, Westaway SK, Levinson B, Johnson MA, Gitschier J, Hayflick SJ. A novel pantothenate kinase gene (PANK2) is defective in Hallervorden-Spatz syndrome. Nat Genet. 2001;28:345 - 9. [PubMed: 11479594]
- Zorzi G, Zibordi F, Chiapparini L, Bertini E, Russo L, Piga A, Longo F, Garavaglia B, Aquino D, Savoiardo M, Solari A, Nardocci N. Iron-related MRI images in patients with pantothenate kinase-associated neurodegeneration (PKAN) treated with deferiprone: results of a phase II pilot trial. Mov Disord. 2011;26:1756 - 9. [PubMed: 21557313]
Chapter Notes
作者
Jason Coryell, MS; Oregon Health & Science University (2004-2007)
Allison Gregory, MS, CGC (2004-present)
Susan J Hayflick, MD (2002-present)
更新史
- 2017 年 8 月 3 日 (sw) 实时发布综述更新
- 2013 年 1 月 31 日(我)全面更新实时发布
- 2010 年 3 月 23 日(我)实时发布综合更新
- 2008 年 1 月 9 日 (sh) 修订:deletion/duplication analysis不再适用于临床
- 2007 年 1 月 8 日(我)实时发布综述更新
- 2004 年 10 月 27 日(我)实时发布综述更新
- 2003 年 3 月 8 日 (sh) 修订:表 4; 参考文献
- 2003 年 2 月 25 日 (sh) 修订:资源
- 2002 年 8 月 13 日(我)实时发布评论
- 2002 年 3 月 29 日 (sh) 初稿提交