
摘要
临床特征 Emanuel 综合征的临床特征是产前和出生后生长发育迟缓, 小头畸形, 肌张力低下,严重的全面发育迟滞,耳部畸形如耳前皮赘或凹陷,腭裂或高腭弓,先天性心脏缺陷,肾脏异常,以及男性生殖系统异常。
诊断/检测 Emanuel 综合征的诊断基于在先证者中检出一条超数衍生22号染色体[der(22)],表现为22q10-22q11和11q23-qter 片段的重复。
处理
对症处理: 通常需要多学科团队的医疗支持;对胃食管反流,营养,肛门闭锁(或狭窄),腹股沟疝,心脏缺陷,腭裂,髋关节发育不良,其他骨骼并发症,听力损失,隐睾和/或小阴茎,屈光不正,斜视或其他眼科问题的常规护理;持续性的身体,职业和语言治疗;(培训)促进交流的其他沟通方式。
并发症预防: 在有儿科麻醉师的机构中施行镇静和/或手术,过程中应注意预防呼吸道并发症。
监测: 根据患者各器官系统的受累程度,按照需要进行随访;定期评估生长发育情况;由临床遗传学家定期评估。遗传咨询
在99%以上的病例中,患有Emanuel 综合征的先证者父母之一为t(11;22)(q23;q11.2)平衡易位的携带者,而且其表型正常。在绝大多数病例中,父母携带者的t(11;22)也是由其父亲或母亲遗传而来。如果先证者的父母之一是平衡性t(11;22)携带者,则其未来怀孕的可能结果包括:正常染色体,超数der(22)综合征,平衡性t(11;22)携带者和自然流产(由于额外的der(22)或另一种减数分裂分离异常所导致)。再发风险根据先证者的母亲或父亲是平衡易位携带者而有所不同。如果在家族中证实了染色体异常,那么就可以对有风险的胎儿进行产前诊断。诊断
提示性临床表现
当出现以下临床表现时应考虑Emanuel 综合征:
- 严重智力残疾
- 小头畸形
- 不能生长发育
- 耳前皮赘或凹陷
- 耳朵异常
- 腭裂或高腭弓
- 小下颌畸形
- 肾脏异常
- 先天性心脏病
男性生殖器异常
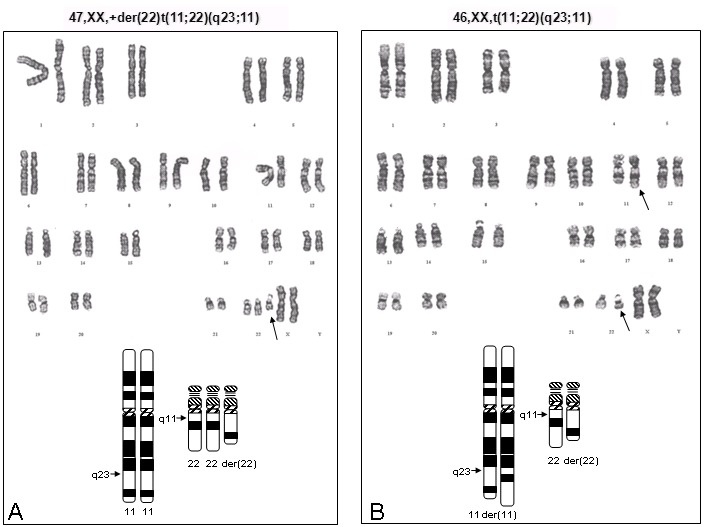
Figure 1
A. 超数der(22)的核型图和模式图, 该超数der(22)导致11q23-qter 的三体和22q cen-q11的三体。 B. 平衡性t(11;22)携带者的核型图和模式图。
检测序列拷贝数的基因组学方法包括核型分析,染色体微阵列(CMA),或通过荧光原位杂交(FISH)检测靶点是否重复。
选择 1
核型分析。 超数der(22) 染色体可通过G显带在500-550条带水平检出。
- (一旦先证者确诊,)应该立即对父母进行核型分析以确定是否为t(11;22)携带者。
罕见的情况下,父母均不是携带者,这时应对先证者行靶向性FISH或CMA对超数衍生染色体进行进一步检测。
选择 2
使用寡核苷酸微阵列的CMA或SNP基因分型微阵列可以对22q近端和11q远端的重复进行检测。
- 应追加核型分析以确定(微阵列技术检测到的)重复是来源于超数染色体,或者:
- 采用来自22q 和11q末端的探针进行FISH检测以确认额外的染色体部分来源于22号染色体,而且易位发生在22q和11q之间。
注意: 染色体微阵列分析不能检测平衡易位,因为其没有遗传物质的增加或丢失。
表 1.
Emanuel 综合征的分子遗传学检测
| 检测方法 | 检测的染色体异常 | 检测敏感性 |
|---|---|---|
| 核型分析 | 超数 der(22) | 100% |
| FISH 1 | 22q11 和 11q23 重复 | 100%(使用11和22探针) |
| CMA 2 | 11号和22号染色体的拷贝数变异 | 100% |
- 1.
.FISH测试使用定位在22q11.2的N25或TUPLE1探针和11q亚端粒探针。在极少数情况下,其中一个亲本不是平衡易位携带者,可用商业化的22q11.2缺失探针和11q的端粒探针对衍生自11号和22号染色体的超数染色体进行鉴定。
- 2.
使用寡核苷酸阵列的染色体微阵列分析(CMA)或SNP微阵列分析。注意:为确定异常的发病机制应采用核型分析或FISH。
临床特点
临床描述
根据支持团体和临床报告,迄今已有超过400例der(22)病例。高死亡率与威胁生命的先天性心脏缺陷,膈疝或肾功能不全等先天畸形有关。出生数月内死亡率最高。随着姑息治疗的改善和时间推移,生存机会得到改善,已有生存至成年的病例。
受累儿童通常于新生儿时期获得确诊,其父母通常为t(11;22)平衡易位携带者。
生长发育 绝大多数病例存在产前和出生后的生长发育迟缓。
颅面部畸形 观察到的异常包括小短头畸形,前额突出,内眦赘皮,下斜睑裂,宽鼻梁、鼻梁凹陷,长而深的人中,小下颌后移(见 图 2).
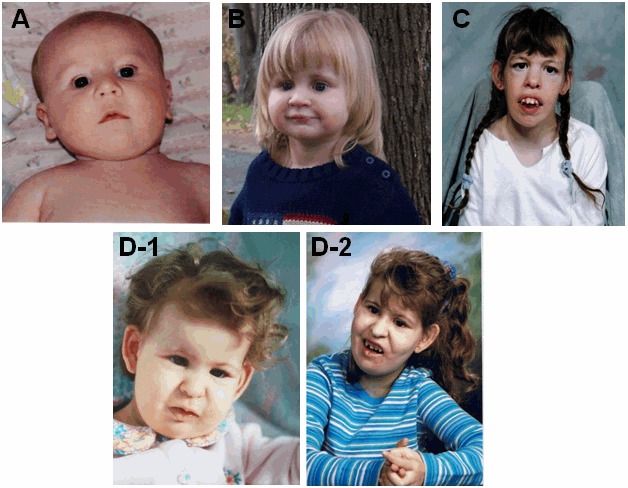
图 2.
四例Emanuel 综合征患者。注意儿童A(约6个月)和B(3岁)中出现的圆脸,眼睛深陷,圆眼睛,前额突出等面部特征。注意病例D的照片分别拍摄于:1岁(D-1)和10岁(D-2),可见随着时间的推移,其面部特征也日益突出。在C(17岁)和D-2图中,两个病例的面部特征非常相似。
外耳廓通常存在畸形,并出现特征性的耳前皮赘或凹陷。已报道可出现严重的小耳畸形合并外耳道闭锁和耳聋。听力损失不常见,但考虑到很难对严重生长发育迟缓的病例进行准确的听力评估,所以有可能低估了轻度听力障碍的比例。
约50%的受累个体存在腭裂,且有报道可见三角嘴凹陷或嘴裂,上颌骨裂、喉软骨软化和鳃窦。此外还可见双歧悬雍垂。
心脏 60% Emanuel 综合征患者可出现先天性心脏缺陷,增加了发病和死亡率。心脏缺陷包括室间隔缺损,房间隔缺损,法洛四联症,永存动脉干,三尖瓣闭锁,主动脉缩窄,异常锁骨下动脉,持续左上腔静脉和动脉导管未闭。
生殖泌尿系统 约30%受累个体可见肾脏畸形,包括肾缺如和不同程度的肾发育不良。男性患者通常还出现
隐睾、小阴囊、小阴茎。女性患者偶尔也可出现子宫畸形。消化道 可见先天性膈疝、膈肌发育不良或膈肌膨出等畸形。
在约20%的受累个体中可见肛门闭锁伴或不伴肛瘘,也观察到没有完全闭锁的肛门狭窄。
腹股沟疝虽不常见,但也有病例报道。
偶尔可观察到胆道闭锁,Hirschsprung病,肝叶异常,肝外胆管,胆囊缺失和体重增加不足常见。体重增加缓慢常见,虽然往往没有描述到具体的喂养问题,但其中常见的有胃食管反流和吸吮和吞咽困难。
肌肉骨骼。所有受累病例都具有明显中枢性肌张力低下。
严重肌张力低下将导致并发症,其中最常见的是脊柱弯曲,并可以导致患者运动发育落后。
常见有骶骨窝。
常见有先天性髋关节脱位或半脱位。
特征性的细长指(趾)和锥形指。
可出现先天性或出生后出现的马蹄内翻足和关节挛缩。
其他较少出现的骨骼系统畸形包括第十三对肋骨,锁骨发育不全,肘外翻,尺桡骨性融合及第四、五脚趾并趾。曾报道1例伴腰脊髓脊膜膨出。
多个病例报告提及存在骨龄延迟。
眼 绝大多数Emanuel 综合征患者视觉正常。眼部异常包括斜视和近视均不常见。上睑下垂和退行性视网膜病变更加少见。
中枢神经系统。 所有患者均有小头畸形。
因为Emanuel综合征的诊断不依赖于大脑影像学检查结果,所以结构性脑异常的发生率尚不清楚,已报告的畸形包括Dandy-Walker畸形,胼胝体发育不全,无鼻无脑畸形,以及嗅球和嗅束缺如。
有报道在少数受累者中出现癫痫发作,另有一部分患者出现异常脑电图但没有临床癫痫发作。
生长发育 所有 Emanuel 综合征儿童均出现严重的全面生长发育迟缓。成人期表现为中重度到重度智力残疾。绝大多数患儿可以在无支持情况下端坐,但由于运动功能不协调,只有一小部分患儿可以学会走路。语言发育明显迟缓,语言接受能力强于语言表达能力,部分患者可以使用简单词汇进行交流。
其他 发现包括先天性免疫球蛋白缺陷、胸腺依赖性免疫缺陷和牙齿发育不良。
基因型-表现型关系
所有Emanuel 综合征患者均存在超数der(22)染色体,该染色体为11q23和22q11发生断裂重接而导致,不同患者的断裂位点几乎发生在同样位置,之间仅相差数个核苷酸[Shaikh et al 1999, Kurahashi et al 2000, Kurahashi & Emanuel 2001]。然而,由于临床表型是与遗传物质的重复有关,目前仍很难确定本病的基因型-表现型关系。尽管每个患者全身各系统受累程度不一样,但大部分患者最终均进展为中重度或重度智力残疾。
表现度
存在超数der(22)染色体的患者是完全显性的。
命名
既往在G显带方法出现前的病例报告中,将这一染色体异常描述为“22号染色体部分三体”或“11号染色体部分三体”。
发病率
超数der(22)染色体是一种罕见的染色体病,其发病率不明。普通人群中的t(11;22)平衡易位携带率也不清楚。
遗传相关疾病
除了在GeneReview 中讨论过的表型,没有与超数der(22)染色体有关的其它已知表型。
在某些研究中发现女性t(11;22)平衡易位携带者绝经前乳腺癌风险升高,然而文献结论并不一致[Jobanputra et al 2005, Wieland et al 2006, Carter et al 2010].
鉴别诊断
下列综合征与Emanuel 综合征有相似的临床表型,染色体核型分析可以确诊Emanuel 综合征并排除其他综合征。
- Pallister-Killian 综合征 (OMIM 601803)
- 其他染色体异常
管理
初步诊断后的评估
关于临床表现能在多大程度上影响发病率和死亡率,目前尚无已发表的指南可以对其进行评估。基于参考文献和作者的经验,建议采用以下检查方法 (如果这些检查还没有完成),进一步确定Emanuel 综合征患者的患病程度以及相应的治疗需求:
- 检查是否有腭裂
- 通过超声心动图检查是否存在心脏缺陷。房间隔缺损是最常见的心脏缺陷,不一定能通过心脏听诊检查发现。
- 通过肾脏超声检查评估肾脏结构畸形;如果有提示话,通过膀胱输尿造影评估膀胱输尿管返流情况。
- 采用合适的放射影像学检查方法评估胃肠道结构畸形,尤其注意肛门狭窄或膈肌异常,胃食道返流等。
- 喂养和吞咽情况评价。
- 采用合适的放射影像学检查方法评估骨骼异常包括髋关节发育不良、关节挛缩、马蹄内翻足,脊柱侧弯和尺桡骨性融合。
- 耳科检查评估外耳道闭锁或狭窄。
- 用听觉脑干反应测试与耳声发射测试进行听力评估 (测试的更多内容见“遗传性听力损失和耳聋”部分)
- 用散瞳眼底内窥镜检查评估视力及斜视。
- 检查男性患者泌尿生殖系统有无隐睾和/或小阴茎。
- 由儿童发育专业的儿科医师和治疗师进行评估并实施以加强沟通能力为主的教育/治疗干预。
- 请临床遗传学家和/或遗传医师会诊,并进行遗传咨询,以及确定存在发病风险的亲属(+der(22)染色体几乎都是遗传自携带者父母)。
对症处理
鉴于Emanuel 综合征患者的年龄和全身各系统的受累程度,需要由多个不同专业的专家联合进行会诊。
对某些严重的结构畸形和/或肾衰竭患者,应采取适当的姑息治疗。
- 对胃食管反流的规范治疗,对于不能生长发育的,补充配方营养并考虑肠内营养。
- 手术治疗肛门闭锁 (或狭窄) 和腹股沟疝。
- 对以下异常采取常规干预措施:
- 心脏缺陷
- 腭裂
- 髋关节发育不良和其他骨骼系统并发症。采用辅助设备如助行器等协助步行。
- 听力损失
- 隐睾和/或小阴茎
- 屈光错误、斜视或其他眼科问题
- 癫痫,如有
- 持续性的身体、职业和言语治疗以改善病情。
- 因为口头语言能力非常有限,应训练其采用其他方法以促进沟通。
预防二次并发症
在镇静和/或手术过程中的护理应由儿科麻醉师负责,因为Emanuel 综合征患儿通常有气道狭小、不同程度的腭畸形和喉软骨软化。
监测
应采取以下措施:
- 基于患者的全身各系统受累程度进行必要的随访。
- 定期评估生长发育进展,以对治疗性干预和教育方式进行指导。
- 临床遗传医师进行周期性评估以告知家庭疾病的新进展和/或建议。
亲属风险的评估
见遗传咨询中的有关风险的评估。
尚在临床试验阶段的治疗方法
搜索 ClinicalTrials.gov 可以获得大量疾病的临床试验的信息。注意:可能没有针对本病的临床试验研究。.
其他
应当告知病人及其家属有关本病的自然史、治疗、遗传模式、其他亲属的遗传风险以及面向消费者的资源。
遗传咨询
遗传咨询的内容是向患者及其家庭提供该病的性质、遗传方式及其可能造成的影响方面的信息,帮助他们做出基于足够背景知识,以及符合个人情况的决定。 以下几个段落是涉及遗传风险评估, 根据家族史和遗传学检测来确定家庭成员的遗传状态。这一段的目的并不是为了解决所有患者可能面临的个人、文化或伦理问题,或者替代遗传学专业人员的咨询工作。-编辑。
遗传模式
Emanuel 综合征是遗传性染色体异常。它是t(11;22)(q23;q11)平衡易位染色体在减数分裂时发生3:1不平衡分离而导致的。这一染色体重排是人类已知唯一的复发性的非罗氏平衡易位。
家庭成员的风险
先证者的父母
建议对父母进行染色体核型分析以检测是否存在t(11;22)平衡易位。
在99%以上病例中,Emanuel 综合征先证者的双亲之一是t(11;22)(q23;q11)平衡易位携带者,并且表型正常。仅在一个病例报道中发现超数der(22)染色体是由于父亲生殖细胞中新发的t(11;22)发生可能的不平衡邻位-1分离,加上母亲减数分裂I时发生22号染色体不分离而导致的。
从统计上看,母亲为t(11;22)平衡易位携带者的几率高于父亲。
绝大多数情况下,携带者父母的t(11;22)也是遗传自其父母之一。
先证者的兄弟姐妹
先证者的兄弟姐妹如无Emanuel 综合征临床表现:
无Emanuel 综合征患病风险;
几乎没有可能携带另一不同的不平衡染色体异常;
有50%的可能性为染色体平衡易位;
有50%的可能性为染色体正常。
先证者的兄弟姐妹如有Emanuel 综合征临床表现(如重度发育迟缓,生长缓慢,多发先天畸形等)几乎都携带超数der(22)染色体。
如果先证者父母之一是t(11;22)携带者,则其将来怀孕时胎儿存在以下几种可能性:
正常染色体
超数der(22)染色体综合征
平衡性t(11;22)携带者
由于超数der(22)或其他减数分裂异常分离方式导致的自然流产
在任一次怀孕中,以上四种可能妊娠结局的发生几率均不能确定。此外根据携带者(是先证者父亲还是母亲)的不同,其风险也不同 [Fraccaro et al 1980, Zackai & Emanuel 1980]:
如果母亲为携带者,则其生育携带超数der(22)的婴儿的风险要高于父亲为携带者的情况。
如果父母之一为携带者,则其生育携带超数der(22)活产婴儿的总体风险介于1.8%和5.6%之间。
如果父母之一为携带者,则其因胎儿因携带超数der(22)或其他减数分裂异常而导致自然流产的总体风险介于23%和37%之间。
先证者的子女 目前没有Emanuel 综合征患者生育的报道。
其他家庭成员
其他家庭成员的患病风险取决于先证者的父母:如果父母之一携带平衡性t(11:22)易位,则其家庭成员可能也携带同样的平衡易位或患有Emanuel综合征。对于家族中的其他携带者,其风险情况与上文讨论过的先证者父母一致。
应对有风险的家庭成员进行染色体分析。
遗传咨询的相关问题
生育规划
怀孕前是遗传风险值评估和产前检查可行性分析的最佳时间。
应当对已知有染色体重排、或有这种风险的年轻成人提供遗传咨询(讨论后代潜在的风险,以及生育的可能方式等)。
产前诊断和胚胎植入前遗传学诊断
如果在患病的家族成员中检出超数der(22),就可以考虑对具有风险的胎儿进行产前诊断,还可以考虑进行植入前诊断。
信息来源
为了该病患者及其家庭成员的方便,GeneReviews的员工已经选择了下述的针对该病的,或者包括其他GeneReviews疾病的患者支持组织和患者注册组织。GeneReviews不为其他组织提供的信息承担责任。选择这些组织的标准,请点击 此处获取详细信息。
- Chromosome 22 Centralc/o Murney Rinholm7108 Partinwood DriveFuquay-Varina NC 27526Phone: 919-567-8167Email: usinfo@c22c.org
- Chromosome 22 Central
分子遗传学
在下面的分子生物学表和OMIM表内的信息可能和GeneReview里其他部分的信息不一致:表格里的信息可能更加新。-编辑.
表 B.
Emanuel综合征 OMIM 词条 (在 OMIM中查看)
| 609029 | EMANUEL 综合征 |
分子遗传发病机制
Emanuel综合征是由于染色体11q和22q片段重复引起的,因为这些片段上包含了数量众多的基因,所以该病的分子遗传发病机制还不清楚。
参考资料
参考文献
- Carter MT, Barrowman NJ, St Pierre SA, Emanuel BS, Boycott KM. Risk of breast cancer not increased in translocation 11;22 carriers: analysis of 80 pedigrees. Am J Med Genet A. 2010;152A:212 - 4. [PMC free article: PMC2802109] [PubMed: 20034094]
- Fraccaro M, Lindsten J, Ford CE, Iselius L. The 11q;22q translocation: a European collaborative analysis of 43 cases. Hum Genet. 1980;56:21 - 51. [PubMed: 7203479]
- Jobanputra V, Chung WK, Hacker AM, Emanuel BS, Warburton D. A unique case of der(11)t(11;22),-22 arising from 3:1 segregation of a maternal t(11;22) in a family with co-segregation of the translocation and breast cancer. Prenat Diagn. 2005;25:683 - 6. [PMC free article: PMC2810961] [PubMed: 16049998]
- Kurahashi H, Emanuel BS. Long AT-rich palindromes and the constitutional t(11;22) breakpoint. Hum Mol Genet. 2001;10:2605 - 17. [PubMed: 11726547]
- Kurahashi H, Shaikh TH, Hu P, Roe BA, Emanuel BS, Budarf ML. Regions of genomic instability on 22q11 and 11q23 as the etiology for the recurrent constitutional t(11;22). Hum Mol Genet. 2000;9:1665 - 70. [PubMed: 10861293]
- Shaikh TH, Budarf ML, Celle L, Zackai EH, Emanuel BS. Clustered 11q23 and 22q11 breakpoints and 3:1 meiotic malsegregation in multiple unrelated t(11;22) families. Am J Hum Genet. 1999;65:1595 - 607. [PMC free article: PMC1288370] [PubMed: 10577913]
- Wieland I, Muschke P, Volleth M, Ropke A, Pelz AF, Stumm M, Wieacker P. High incidence of familial breast cancer segregates with constitutional t(11;22)(q23;q11). Genes Chromosomes Cancer. 2006;45:945 - 9. [PubMed: 16845657]
- Zackai EH, Emanuel BS. Site-specific reciprocal translocation, t(11;22) (q23;q11), in several unrelated families with 3:1 meiotic disjunction. Am J Med Genet. 1980;7:507 - 21. [PubMed: 7211960]
注解
作者注解
致谢
我们感谢Stephanie St Pierre, Chromosome 22 Central 以及病患家庭对我们研究工作的帮助与支持.
更新历史
- 31 August 2017 (sw) 系统性更新发布
- 5 February 2015 (me) 系统性更新发布
- 11 May 2010 (me) 系统性更新发布
- 20 April 2007 (me) 内容发布
- 8 January 2007 (bse) 初始稿件