
简介
临床特征 Fukuyama先天的肌营养不良症(FCMD)的特征在于肌张力低下,全身性对称性肌肉无力和CNS发育障碍,导致鹅卵石性脑小脑畸形,伴大脑和小脑皮质发育不良。可以识别出轻度,典型和严重的表型。发作通常发生在婴儿早期,吮吸能力较弱,哭声较弱和易激惹。患儿臀部,膝盖和指间关节都有挛缩。后来的特征包括肌病性面部外观,小腿和前臂的假性肥大,运动和言语延迟,智力障碍,癫痫发作,眼科异常(包括视力障碍和视网膜发育异常)以及十岁后进行性心脏受累。吞咽障碍发生在患有严重FCMD的个体和年龄超过10岁的个体中,导致反复吸入性肺炎和死亡。
诊断/测试 FCMD的诊断是在先证者中通过分子遗传学检测鉴定FKTN中双等位基因的致病变异而建立的。
管理 表型治疗:物理治疗和伸展运动,骨科并发症的治疗,长腿矫正器和轮椅等辅助设备,使用无创呼吸道辅助工具或气管切开术,急性呼吸道感染的及时治疗,抗癫痫药,药物和/或手术治疗对于胃食管反流,胃造口管放置以确保足够的热量摄入时,按照心脏病专家的要求进行心肌病治疗。
监测:监视:
- 晚期疾病患者的呼吸功能;
- 对于年龄超过10岁的个人,通过胸部X光,心电图和超声心动图检查是否有心肌受累;
- 胃肠道功能,胃食管反流的体征/症状;
- 足部畸形和脊柱侧弯。
遗传咨询 FCMD以常染色体隐性遗传的方式遗传。 受累的患者的每个同胞都有25%的机会发病,有50%的机会成为无症状的携带者,有25%的机会不受影响。如果已知家族中的致病变异,则可以对高危家庭成员进行携带者测试,并针对高危妊娠进行产前诊断。
诊断
提示性发现
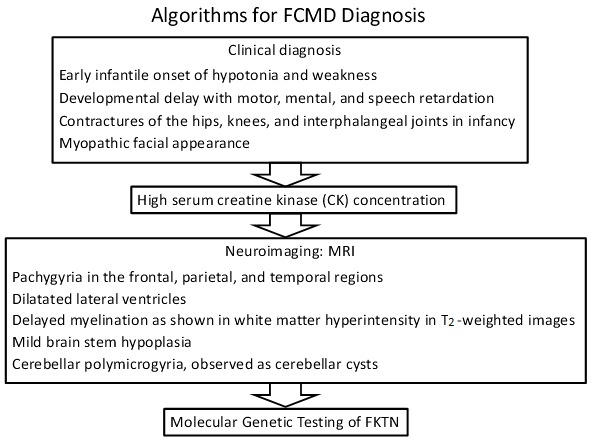
具有以下临床,影像学,实验室检查和组织病理学发现的个人应怀疑 Fukuyama 先天的肌营养不良。
临床发现
- 婴儿早期出现肌张力低下和无力,臀部,膝盖和指间关节挛缩(100%的个体)
- 严重的运动和言语延迟以及智力障碍,尚有部分社交技能(100%)
- 僵硬开始于儿童早期,然后是弥漫性和广泛性肌肉消瘦(最明显的是近端),后来进行性关节挛缩(100%)
- 肌病性面部外观(100%)
- 婴儿后期小腿和前臂的假性肥大(50%)
- 癫痫发作(无热和/或发热)(50%)
- 眼科异常,包括53%的视力障碍和32%的视网膜异常[Saito & Kobayashi 2001]。存在时视网膜异常轻度和局灶。视网膜发育不良是一种病理学诊断,其依据是发现未成熟感光细胞的玫瑰花结。
- 家族史与常染色体隐性遗传一致
神经影像学发现。 MRI揭示了鹅卵石小脑畸形的发现,包括以下五个主要异常:
- 脑表面不规则或有卵石样改变;在额叶,顶叶和颞叶区域具有宽皮层(厚皮层)的宽回回;有时是类似于多小脑回的小型和不规则回旋区域
- 侧脑室扩张
- 在T2加权图像上出现高强度的白质异常,在T1加权图像上出现低强度的白质异常[Kato et al 2000]表示髓鞘形成延迟了 [Kato et al 2006, Kato et al 2010]而不是髓鞘异常
- 有些人脑干轻度发育不良
- 小脑回多和小脑囊肿(23/25个人 [Aida et al 1994])
此外:
- 皮质的厚度通常不超过约1厘米。
- 脑盖发育不佳,留下Sylvian裂缝。
实验室检查结果 血清肌酸激酶(CK)浓度:
- 年龄<6岁:比正常人高10-60倍
- 年龄≥7岁:比正常高5-20倍
- 卧床不起的人:正常
组织病理学 肌肉活检:
- 发现是肌营养不良的特征。主要特征是无肌肉变性和再生的间质纤维化,这使Fukuyama先天的肌营养不良症与 Duchenne muscular dystrophy有所区别 [Taniguchi et al 2006].
- 使用α-dystroglycan抗体的免疫组织化学染色显示骨骼肌表面膜上选择性缺乏α-dystroglycan[Hayashi et al 2001].
注意:随着分子遗传学检测的发展,不再需要进行肌肉活检来确定FCMD的诊断。
肌电图表现是肌营养不良的特征。
建立诊断
通过在先症者中通过分子遗传学检测鉴定FKTN中双等位基因的致病变异,可以确定 Fukuyama先天的肌营养不良症的诊断(参见 Table 1和 Figure 1)
分子遗传学检测方法可以包括靶向基因-的检测(单基因检测,multigene panel)和综合基因组的检测(外显子组测序, 基因组测序)的组合,具体取决于表型。
以基因为目标的测试要求临床医生确定可能涉及哪些基因,而基因组的测试则不需要。因为FCMD的表型很宽,具有“Suggestive Findings ”中所述独特发现的个体很可能使用基因靶向检测来诊断(参见Option 1),而具有与许多其他遗传性肌营养不良性疾病没有区别的表型的个体则更多。可能是通过基因组测试诊断出来的(请参阅 Option 2)。
选项1
当表型和实验室发现提示FCMD的诊断时,分子遗传学检测方法可以包括单基因检测或multigene panel使用:
- 单基因测试 对于日裔,韩裔和/或中国血统的个人,请首先对c.*4392_*4393insAB185332.1 建立者变异进行针对性分析。如果仅识别出一个或没有一个致病性变异,则对整个基因进行序列分析。
注意:对于具有朝鲜血统的人,如果仅识别出一个或没有致病性变异,请考虑进行 序列分析以检测朝鲜族建立者变异 c.647+2084G>T。
- 包含FKTN和其他感兴趣的基因的肌营养不良症multigene panel (参见 Differential Diagnosis)最有可能以最合理的代价鉴定出该病的遗传原因,同时限制了意义不确定和无法解释表型变异出现。注意:(1)套餐中包含的基因和每个基因所用测试的诊断 敏感性因实验室而异,并且可能随时间而变化。 (2)一些套餐可能包含与本 GeneReview中讨论的病症无关的基因。 (3)在某些实验室中,套餐选项可能包括实验室定制的设计套餐和/或定制的以表型为重点的外显子组分析,其中包括临床医生指定的基因。 (4)套餐中使用的方法可能包括序列分析, deletion/duplication analysis和/或其他非基于序列的测试。
有关多基因套餐的介绍,请单击here。有关订购基因检测的临床医生的更多详细信息,请参见here。
选项2
当该表型与其他许多以肌营养不良为特征的遗传性疾病没有区别时,综合基因组的检测(不需要临床医生确定可能涉及哪些基因)是最佳选择。外显子组测序是最常用的方法;基因组测序也是可能的。
有关全面的基因组的测试的介绍,请单击here。可以在here找到有关订购基因组测试的临床医生的更多详细信息。
Table
Fukuyama先天性肌营养不良症的分子遗传学检测
| 基因1 | 方法 | 致病变异的比例2 通过方法检测的种族 | |||
|---|---|---|---|---|---|
| 日本 | 非日裔亚洲人 | 非亚裔 | |||
| FKTN | 靶向分析 | c.*4392_*4393insAB185332.1 | 98% 3 | 77% 4, 5 | 0% |
| c.647+2084G>A | 8% | 38% (Korean) | 0% | ||
| c.139C>T | 7% | 60% (Chinese) | Rare | ||
| 测序分析 6 | 8% 7 | 8% | 100% 8 | ||
| 基因靶向 deletion/duplication analysis 9 | Rare | Rare | Rare | ||
- 1.
染色体 位点和蛋白质信息见 Table A. Genes and Database。
- 2.
有关在该基因中检测到的等位基因变异的信息见 Molecular Genetics
- 3.
在对107名患有FCMD的日本人进行的分析中:80(75%)人是 建立者变异的纯合性; 25种(23%)是 c.*4392_*4393insAB185332.1的复合杂合,包括九种(8%)的c.647+2084G>T 和七种(7%)的 c.139C>T [Kobayashi et al 2017]。
- 4.
在对13名韩国FCMD个体的分析中:c。* 4392_ * 4393insAB185332.1变体中有3个(23%)是纯合性;七个(54%)是c.*4392_*4393insAB185332.1的复合杂合,包括五个(38%)的 c.647+2084G>T [Lim et al 2010]。
- 5.
- 6.
- 7.
序列分析无法确定在日本血统的个人中最常见的建立者变异,c.*4392_*4393insAB185332.1。
- 8.
包括鉴定携带者频率为0.7%(2/299个人)的 Ashkenazi Jewish 建立者变异 c.1167_1168insA [Chang et al 2009].。
- 9.
基因靶向的deletion/duplication analysis基因内的缺失或重复。所使用的方法可能包括 quantitative PCR,远程PCR,多重连接依赖探针扩增(MLPA),以及旨在检测单外显子缺失或重复的 基因靶向微阵列。
临床特征
Fukuyama 先天的肌营养不良症(FCMD)的特征在于骨骼肌营养不良,以及CNS迁移发育障碍,大脑和小脑皮质发育异常。临床特征是肌张力低下,虚弱和精神运动迟钝。可以识别出轻度,典型和严重的表型。表型谱的范围从重度表型末期的Walker-Warburg综合征(WWS)型[Manzini et al 2008, Chang et al 2009, Yis et al 2011]到温和的肢带型肌营养不良表型 [Puckett et al 2009, Yis et al 2011, Fiorillo et al 2013].
疾病发作通常发生在婴儿早期。最初的症状包括吮吸不佳,哭声小,易激惹和运动发育延迟。存在对称的全身性肌肉无力和肌张力低下。一些婴儿体重增加慢。
主要的近端肌张力减退表现为肩膀和躯干的过度伸展。还观察到髋关节伸展,髋外展和膝关节伸展的局限性,并随时间增加。在婴儿后期,小腿和前臂的“浮肿的”脸颊和假肥大明显。肌肉坚硬,具有纤维质地。婴儿早期后,深层肌腱反射减弱或消失。面部肌肉受累(肌无力相)在6至12个月时就很明显,并且随着年龄的增长而增加[Osawa et al 1997]。在儿童时期,张口,下颌前突和大舌症变得更加明显。六岁以后会出现吞咽困难。
发育迟缓和言语迟缓发生在所有患者中。智商范围通常为30到60。在轻度FCMD患者中,智商大于35。在患有严重FCMD的人中,智商低于30。典型FCMD的人的最佳状态包括数十个口头单词,无助地坐着以及沿着臀部的地板滑动。轻度FCMD的患者可以独立行走或站立。患有严重FCMD的患者可能缺乏头部控制或独立坐下的能力。
具有FCMD的人的社交能力不如躯体和智力的严重影响 [Saito & Kobayashi 2001]。患有FCMD的儿童往往是他们的托儿所,幼儿园或小学的最爱。甚至患有FCMD的 受累的严重的人也表现出眼神交流,能认出家人并通过发声提出要求。没有观察到自闭症特征。
癫痫 超过60%的受累的患者会发生癫痫发作 [Yoshioka et al 2008]。对于日本 建立者变异 (c.*4392_*4393insAB185332.1), 纯合性个体的高热和无热惊厥发作的平均年龄分别为5.4岁和4.6岁。在日本建立者变异和其他致病性变异的复合杂合个体中,高热和无热惊厥发作的平均年龄分别为3.6和3.7岁[Yoshioka et al 2008]。
眼部异常包括40%-53%的个体的屈光不正(近视和远视)。 FCMD较严重者中视网膜异常可见32%[Chijiiwa et al 1983, Tsutsumi et al 1989, Osawa et al 1997]。然而,视网膜发育不良是轻度和局灶性的。
在通过分子遗传学检测确诊的严重FCMD的一些个体中,严重的眼部异常包括小眼症,视网膜脱离,视网膜发育不全,白内障和青光眼 [Mishima et al 1985, Hino et al 2001, Saito & Kobayashi 2001, Manzini et al 2008, Chang et al 2009]。值得注意的是,FCMD中不存在肌肉眼脑疾病(MEBD)或WWS的特征性眼部发现(例如,前房异常,青光眼)。
缓慢进行性心脏受累是FCMD的特征。心脏功能障碍的临床进展明显轻于Duchenne muscular dystrophy(杜氏肌营养不良症,DMD)[Yamamoto et al 2017]。死后的发现证明,寿命超过十年的个体倾向于发生心肌纤维化[Finsterer et al 2010]。在34位FCMD患者中,使用M型和多普勒超声心动图评估左心室(LV)功能时,年龄15岁以上的11位患者中有8位显示左室收缩功能降低[Nakanishi et al 2006]。脑钠肽浓度与年龄或左心室射血分数无相关性[Yamamoto et al 2017].
吞咽功能障碍 婴儿FCMD(特别是严重的FCMD)的个体以及年龄超过十岁的晚期疾病的个体均发现吞咽功能障碍。无法吞咽会导致反复吸入性肺炎和死亡[Hill et al 2004].
Murakami et al [2012] 报道在病毒性感染高热发作几天后,肌肉无力突然加重,血清肌酸激酶(CK)和尿肌红蛋白水平明显升高,偶而导致死亡。
神经病理学 对FCMD的大脑检查显示神经元迁移缺陷引起的鹅卵石性脑畸形,伴有脑和小脑皮质发育异常有关的变化[Saito et al 2000]。这些变化与MEBD和WWS中描述的异常相似,但严重程度通常不如MEBD和WWS中描述的异常。
婴儿可在大脑半球表面显示广泛的巨脑回畸形,这一特征在额叶尤其是颞叶比顶叶和枕叶更突出。经常在顶枕叶的皮质表面上发现多小脑回的变体(请参阅Polymicrogyria Overview)。
在畸形的小脑皮层或多小脑回区域下方观察到了排列有分子层并包含软脑膜组织的小脑囊肿 [Aida 1998]。尽管这些变化足以诊断出鹅卵石性脑病,但这些变化并未区分FCMD和其他引起MEBD或WWS的原因。
在少年和成年病例中,回旋区较集中,并局限于枕叶。畸形皮层的中脑或回旋区可能与多小回旋区交替,这是由于回旋融合和神经胶质间充质组织过度迁移进入蛛网膜下腔所致。
观察到由继发性发育不全引起的髓质畸形或平坦的延髓 ,伴有小的基础桥和脊髓凹槽[Saito & Kobayashi 2001].
在胎儿的情况下,神经元和神经胶质细胞通过神经胶质局限性局灶性缺损迁移,形成疣状结节,这是皮质发育异常的最初表现。因此,中枢神经系统实质转移到蛛网膜下腔是一个病理过程,被认为对皮层发育异常的发展至关重要。
基因型-表型相关
Kondo-Iida et al [1999]和Kobayashi et al [2017]分析了107名无亲属的 受累的患者的FKTN。纯合性日本 建立者变异 c.*4392_*4393insAB185332.1的个体显示的 表型比具有这种致病性变异与其他 等位基因的致病性错义或nonsense变异的复合杂合体的表型温和。
严重的表型,包括脑积水和小眼症等WWS样表现,在单碱基变异的复合杂合和日本 建立者变异 (c.*4392_*4393insAB185332.1) 比具有建立者变异的先证者更多见[Yoshioka 2009, Kobayashi et al 2017] 。
Chang et al [2009]在四个具有WWS特征的个体中鉴定出纯合性 c.1167_1168insAFKTN 致病性变异。
Godfrey et al [2006], Godfrey et al [2007], Puckett et al [2009], Yis et al [2011]和 Fiorillo et al [2013] 报告了杂合的个体中轻度LGMD表型ju yo错义变异/移码变异和纯合性致病性错义变异(请参阅Genetically Related Disorders)。
患病率
在日本所有儿童进行性肌营养不良的亚型中,FCMD的患病率仅次于DMD,每10,000例新生儿中的发病率为0.7-1.2。 带有FKTN日本人建立者变异 c.*4392_*4393insAB185332.1源自一个祖先创始人,他的祖先生活在2,000-2,500年前。 在不相关的健康个体中,仅在176条染色体中发现了一条[Kobayashi et al 1998]。
在日本各个地区发现的杂合的携带者平均发生率为188分之一。但是,在朝鲜族人群中,在935个个体中发现了一个携带者,研究人员在203个蒙古族中和来自中国大陆的766个人没有发现任何杂合的致病变异 [Watanabe et al 2005].
FCMD是泛民族,但最常见于具有日本血统的人。
遗传相关(等位基因)疾病
Walker-Warburg综合征(WWS) FKTN中的致病变异也可能与WWS相关。 WWS是肌营养不良症的三种主要表型之一(请参阅 Differential Diagnosis)。
Table 2总结了FKTN中与胚系致病变异相关的其他表型。
鉴别诊断
Fukuyama先天的肌营养不良症(FCMD)是先天性肌营养不良症之一,是临床上和遗传上异质性肌群疾病,其特征是出生时或婴儿早期出现肌无力。先天性肌营养不良(CMD)的主要亚型为层粘连蛋白α-2(肌球蛋白)缺乏症(MDC1A),缺乏胶原蛋白VI的CMD,α-营养不良性蛋白聚糖肌病(由POMT1,POMT2,POMGNT1,FKTN,FKRP,LARGE1, ISPD,POMGNT2,DAG1,TMEM5,B3GALNT2,POMK,B4GAT1和GMPPB)[Godfrey et al 2007, Godfrey et al 2011, Devisme et al 2012, Lim et al 2013, Kang et al 2015, Bouchet-Séraphin et al 2016, Taniguchi-Ikeda et al 2016](请参阅Table 3),SELENON(以前称为SEPN1)相关的CMD(以前称为强直性脊柱综合征或RSMD1)和LMNA相关的CMD(L-CMD)。
α-营养不良患者的三种主要表型为FCMD,Walker-Warburg综合征(WWS)和 肌-眼-脑病(MEBD)[Taniguchi et al 2003, Voglmeir et al 2011, Carss et al 2013]。 α-肌营养不良症的特征是先天的肌营养不良症,伴有特征性脑畸形(鹅卵石[II型]小脑畸形和小脑畸形),眼畸形(典型地涉及视网膜),严重的智力障碍和早期死亡。 FCMD比WWS和MEBD轻,尤其是在脑部和眼科受累方面[Bouchet-Séraphin et al 2016, Taniguchi-Ikeda et al 2016](见 Table 2)。 α-营养不良性蛋白聚糖肌病以 常染色体隐性遗传的方式遗传。
Table 3
α-营养不良性蛋白聚糖肌病的主要表型:FCMD,MEBD和WWS的鉴别
| 表型 | 基因(s) | 表型严重程度 | Brain MRI | |||||
|---|---|---|---|---|---|---|---|---|
| MD | Eye | ID | 脑干 | 小脑 | 小脑囊肿 | 脑积水 | ||
| Fukuyama CMD | FKTN | 中度到重度 | 轻度 | 中度 | 通常正常; 在极少数情况下,发育不良 | 通常正常; 偶尔很小 | 可观察到 | 少见 |
Muscle-eye-brain disease | DAG1 1 GMPPB 2 LARGE1 3 POMGNT1 4 POMT1 POMT2 | 轻度 | 严重 5 | 严重 | 几乎总是很小 | 总是很小 | 可观察到 | 常见 |
| Walker-Warburg syndrome (see OMIM PS236670) | B3GALNT2 6 B4GAT1 7 CRPPA (ISPD) 8 DAG1 9 FKRP 10 FKTN GMPPB 2 LARGE1 3 POMGNT1 4 POMGNT2 11 POMK 12 POMT1 13 POMT2 14 RXYLT1 (TMEM5) 15 | 轻度 | 严重 16 | 严重 | 非常小&扭结在 | 总是很小 | 可观察到 | 几乎所有 |
ID =智力障碍; MD =肌肉营养不良
- 1.
- 2.
- 3.
- 4.
- 5.
严重先天的近视,先天性青光眼,视盘苍白,视网膜发育不全
- 6.
- 7.
- 8.
CRPPA(ISPD)突变会导致严重的WWS,但也是LGMD等较轻表型的原因 [Cirak et al 2013].
- 9.
- 10.
- 11.
- 12.
- 13.
Kang et al 2015
- 14.
- 15.
- 16.
小眼症,视网膜脱离,视网膜发育不全,前房畸形,白内障
管理
关于先天的肌营养不良症(CMD)护理标准的共识性声明已经发表[Wang et al 2010] (full text). Kang et al [2015] (full text)还发布了基于证据的指南,包括针对CMD个人的管理指南。
初步诊断后的评估
为了确定诊断为Fukuyama先天的肌营养不良症(FCMD)的患者的疾病程度和需求,建议进行以下评估(如果诊断未进行的):
- 神经系统评估,包括脑电图和脑部MRI
- 发育评估,包括运动技能,认知和言语评估
- 关节运动范围的物理治疗评估
- 眼科评估
- 缺乏头部控制或无支撑坐下能力的人的进食和吞咽评估
- 热量摄入和营养状况评估
- 咨询临床遗传学家和/或遗传咨询师
表现治疗
没有针对FCMD的治疗方法。适当的多学科管理可以延长FCMD患者的生存期并改善其生活质量。
治疗包括以下内容:
- 物理疗法和伸展运动可促进活动能力和预防挛缩
- 当存在脊柱侧弯时,进行脊柱融合以保持呼吸功能并改善坐姿平衡[Takaso et al 2010, Hino et al 2017, Saito et al 2017]
- 使用机械辅助工具,例如长腿矫正器以保持站立姿势,轮椅帮助行动不便
- 指示时使用呼吸辅助工具,如鼻间歇性正压通气机[Sato et al 2016]
- 注意:提供无创通气,尤其是在夜间,在呼吸窘迫加剧之前。
- 及时治疗急性呼吸道感染;尤其重要,因为这些感染是FCMD患者住院和死亡的最常见原因
- 必要时使用抗癫痫药
- 胃食管反流的医学和/或外科治疗
- 胃造口管放置时,以确保足够的热量摄入
- 心脏病专家对心肌病的治疗
监视
以下基于证据指南的监测 [Kang et al 2015]旨在促进生长和发育,减轻合并症,优化功能并减低死亡率,同时使生活质量最大化:
- 至少每三个月发作一次且每六个月发作一次脑电图的患者的临床评估
- 监测年龄超过10岁的晚期FCMD患者的呼吸功能。那些存活超过20岁的人可能需要气管切开术或无创呼吸支持。
- 通过胸部X光,心电图和超声心动图监测年龄大于10岁的个体的心肌受累情况
- 由合格的专家通过视频荧光吞咽评估,上消化道图像和pH监测器对胃肠道功能进行观察/评估,以评估胃食管反流
- 监测足部畸形和脊柱侧弯
评估处于危险中亲戚
有关为遗传咨询目的而对高危亲戚进行测试的相关问题,请参见 Genetic Counseling 。
正在调查的疗法
反义寡核苷酸治疗 Taniguchi-Ikeda et al [2011]报告说,在具有FCMD的个体的细胞培养物中和在模型小鼠中引入靶向反义寡核苷酸可以恢复正常的果胶素mRNA表达和蛋白质产生。他们的工作证明了剪接调节疗法有望成为FCMD的一种基本临床疗法。
补充疗法 Kanagawa et al [2016] 报告说,核糖5-磷酸是哺乳动物中的一个功能性聚糖单元,其翻译后修饰途径中的缺陷是引起ISPD-,FKRP-和FKTN-α-营养不良的原因。由于ISPD缺乏会导致细胞CDP-核糖醇的丧失或严重减少,因此补充CDP-核糖醇可能有效治疗FCMD。 Gerin et al [2016] 表明,向具有ISPD致病性变异的个体的成纤维细胞补充核糖醇可部分拯救α-营养不良糖基化。
基因治疗 分别使用重组腺相关病毒血清型9介导的fukutin-和FKRP基因的导入小鼠FCMD和LGMD模型 [Kanagawa et al 2013, Xu et al 2013]。早期干预将是高度优先的选择 [Vannoy et al 2017].
在美国搜索ClinicalTrials.gov ,在EU Clinical Trials Register,以获取有关多种疾病和状况的临床研究信息。
遗传咨询
遗传咨询是为个人和家庭提供有关遗传疾病的性质,遗传和影响的信息,以帮助他们做出明智的医疗和个人决定的过程。下一节讨论遗传风险评估以及家族史和基因检测的使用,以阐明家族成员的遗传状况。本部分的目的不是要解决个人可能面临的所有个人,文化或伦理问题,也不能替代遗传专家的咨询。 —编者。
遗传方式
Fukuyama先天的肌肉营养不良(FCMD)以常染色体隐性遗传的方式遗传。
家庭成员的风险
先证者的父母
先证者的同胞
先证者的后代 患有FCMD的个体不会繁殖。
其他家庭成员 先证者's父母的每个同胞有50%FKTN致病性变异 携带者的风险。
携带者(杂合子)检测
对高危亲属进行携带者检测需要事先鉴定该家族中的FKTN致病变异。
相关的遗传咨询问题
家庭计划
DNA库是DNA(通常从白细胞中提取)的存储,以备将来使用。因为将来测试方法和我们对基因,等位基因变异和疾病的理解可能会有所改善,所以应考虑受累的患 DNA库。
产前检测和植入前遗传学诊断
资源
GeneReviews工作人员选择了以下特定疾病和/或综合保护组织和/或登记册,以保护患有该疾病的个人及其家人。 GeneReviews对其他组织提供的信息概不负责。 有关选择标准的信息,请单击here.
- Cure CMD19401 South Vermont AvenueSuite J100Torrance CA 90502Phone: 323-250-2399Fax: 310-684-2023Email: info@curecmd.org
- European Neuromuscular Centre (ENMC)Lt Gen van Heutszlaan 63743 JN BaarnNetherlandsPhone: 31 35 5480481Fax: 31 35 5480499Email: enmc@enmc.org
- Japan Muscular Dystrophy Association3-43-11 Fukushi Zaidan BldgTokyo 170-0005JapanPhone: 81-3-6907-3521
- Muscular Dystrophy Association - USA (MDA)222 South Riverside PlazaSuite 1500Chicago IL 60606Phone: 800-572-1717Email: mda@mdausa.org
- Muscular Dystrophy UK61A Great Suffolk StreetLondon SE1 0BUUnited KingdomPhone: 0800 652 6352 (toll-free); 020 7803 4800Email: info@musculardystrophyuk.org
- Congenital Muscle Disease International Registry (CMDIR)The CMDIR is a patient self-report registry with the goal to register the global 先天的 muscle disease population including persons with congenital myopathy, congenital muscular dystrophy, and congenital myasthenic syndrome. The CMDIR registers 受累的 individuals of all ages with symptoms from birth through late onset (limb-girdle). Registrants will receive educational information and connections to others in the CMD community, and will be contacted about potential participation in clinical trials for their CMD subtype.19401 South Vermont AvenueSuite J100Torrance CA 90502Phone: 323-250-2399Fax: 310-684-2023Email: counselor@cmdir.org; sarah.foye@cmdir.org
by分子遗传
分子遗传学和OMIM表中的信息可能不同于GeneReview中的其他信息:表中可能包含最新信息. —编者.
Table A
Fukuyama先天性肌营养不良:基因和数据库
Table B
Fukuyama先天性肌营养不良OMIM 条目 (View All in OMIM)
分子发病机理
介绍 肌营养不良症基因FKTN,FKRP和ISPD编码该结构合成所必需的酶:fukutin蛋白和FKRP将核糖醇磷酸转移到α-营养不良糖的糖链上,ISPD合成CDP-核糖醇,这是fukutin蛋白和FKRP的供体[Kanagawa et al 2016]。 Fukutin,FKRP和ISPD直接参与串联RboP的合成:fukutin和FKRP是RboP转移酶,ISPD参与细胞CDP-核糖醇的合成 [Kanagawa et al 2016]。 此外,TMEM5是用于修饰核糖醇的UDP-木糖基转移酶,该磷酸二酯与α-营养不良聚糖上的核心M3聚糖有磷酸二酯键 [Praissman et al 2016]。
病因的机制 功能丧失, FCMD是核糖磷酸缺乏症。
Table 4
值得关注的FKTN致病变异
| 参考序列 | DNA核苷酸变化 | 预测的蛋白质变化 | 评论[参考] |
|---|---|---|---|
| NM_001079802 NP_001073270 | c.1167_1168insA | p.Phe390IlefsTer14 | 建立者变异 Ashkenazi Jewish [Chang et al 2009] |
| NM_006731 | c.647+2084G>T 1 | p.Arg216SerfsTer10 | 韩国建立者变异 [Lim et al 2010]; 日本第二大常见变异 [Kobayashi et al 2017] |
| NM_006731 NP_006722 | c.*4392_*4393insAB185332.1 2, 3 | 日本建立者变异 [Kobayashi et al 1998] | |
| NM_001079802 | c.139C>T | p.Arg47Ter | 日本第三大常见变异 [Kobayashi et al 2017] |
表中列出的变异由作者提供。 GeneReviews员工尚未独立验证变异的分类。
GeneReviews遵循人类基因组变异学会(varnomen
- .hgvs.org )的标准命名约定。 有关命名法的说明,请参见Quick Reference。- 1.
激活假外显子的内含子的深内含子的变异
- 2.
在3'非翻译区串联重复序列的3 kb反转录转座子插入 [Kato et al 2004]。 星号表示3'UTR中的变异; 该数字表示终止密码子之外的核苷酸位置。AB185332
- .1 是插入的反转录转座子序列的登录号。- 3.
也称为NM_001079802
- .1 :c。* 4392_4393ins3062
参考文献
发布的准则/共识声明
- Kang PB, Morrison L, Iannaccone ST, Graham RJ, Bönnemann CG, Rutkowski A, Hornyak J, Wang CH, North K, Oskoui M, Getchius TS, Cox JA, Hagen EE, Gronseth G, Griggs RC; Guideline Development Subcommittee of the American Academy of Neurology and the Practice Issues Review Panel of the American Association of Neuromuscular & Electrodiagnostic Medicine. Evidence-based guideline summary: evaluation, diagnosis, and management of congenital muscular dystrophy. Available online. 2015. Accessed 6-27-19.
- Wang CH, Bonnemann CG, Rutkowski A, Sejersen T, Bellini J, Battista V, Florence JM, Schara U, Schuler PM, Wahbi K, Aloysius A, Bash RO, Béroud C, Bertini E, Bushby K, Cohn RD, Connolly AM, Deconinck N, Desguerre I, Eagle M, Estournet-Mathiaud B, Ferreiro A, Fujak A, Goemans N, Iannaccone ST, Jouinot P, Main M, Melacini P, Mueller-Felber W, Muntoni F, Nelson LL, Rahbek J, Quijano-Roy S, Sewry C, Storhaug K, Simonds A, Tseng B, Vajsar J, Vianello A, Zeller R; International Standard of Care Committee for Congenital Muscular Dystrophy. Consensus statement on standard of care for congenital muscular dystrophies. Available online. 2010. Accessed 6-27-19. [PMC free article: PMC5207780] [PubMed: 21078917]
引用文献
- Aida N. Fukuyama congenital muscular dystrophy: a neuroradiologic review. J Magn Reson Imaging. 1998;8:317 - 26. [PubMed: 9562058]
- Aida N, Yagishita A, Takada K, Katsumata Y. Cerebellar MR in Fukuyama congenital muscular dystrophy: polymicrogyria with cystic lesions. AJNR Am J Neuroradiol. 1994;15:1755 - 9. [PubMed: 7847224]
- Astrea G, Romano A, Angelini C, Antozzi CG, Barresi R, Battini R, Battisti C, Bertini E, Bruno C, Cassandrini D, Fanin M, Fattori F, Fiorillo C, Guerrini R, Maggi L, Mercuri E, Morani F, Mora M, Moro F, Pezzini I, Picillo E, Pinelli M, Politano L, Rubegni A, Sanseverino W, Savarese M, Striano P, Torella A, Trevisan CP, Trovato R, Zaraieva I, Muntoni F, Nigro V, D'Amico A, Santorelli FM, et al. Broad phenotypic spectrum and genotype-phenotype correlations in GMPPB-related dystroglycanopathies: an Italian cross-sectional study. Orphanet J Rare Dis. 2018;26;13:170. [PMC free article: PMC6158856] [PubMed: 30257713]
- Bouchet-Séraphin C, Chelbi-Viallon M, Vuillaumier-Barrot S, Seta N. Genes of alpha-dystroglycanopathies in 2016. Article in French. Med Sci (Paris). 2016;32:40 - 5. [PubMed: 27869076]
- Buysse K, Riemersma M, Powell G, van Reeuwijk J, Chitayat D, Roscioli T, Kamsteeg EJ, van den Elzen C, van Beusekom E, Blaser S, Babul-Hirji R, Halliday W, Wright GJ, Stemple DL, Lin YY, Lefeber DJ, van Bokhoven H. Missense mutations in beta-1,3-N-acetylglucosaminyltransferase 1 (B3GNT1) cause Walker-Warburg syndrome. Hum Mol Genet. 2013;22:1746 - 54. [PMC free article: PMC3613162] [PubMed: 23359570]
- Carss KJ, Stevens E, Foley AR, Cirak S, Riemersma M, Torelli S, Hoischen A, Willer T, van Scherpenzeel M, Moore SA, Messina S, Bertini E, Bönnemann CG, Abdenur JE, Grosmann CM, Kesari A, Punetha J, Quinlivan R, Waddell LB, Young HK, Wraige E, Yau S, Brodd L, Feng L, Sewry C, MacArthur DG, North KN, Hoffman E, Stemple DL, Hurles ME, van Bokhoven H, Campbell KP, Lefeber DJ, Lin YY, Muntoni F, et al. Mutations in GDP-mannose pyrophosphorylase B cause congenital and limb-girdle muscular dystrophies associated with hypoglycosylation of alpha-dystroglycan. Am J Hum Genet. 2013;93:29 - 41. [PMC free article: PMC3710768] [PubMed: 23768512]
- Chang W, Winder TL, LeDuc CA, Simpson LL, Millar WS, Dungan J, Ginsberg N, Plaga S, Moore SA, Chung WK. Founder fukutin mutation causes Walker-Warburg syndrome in four Ashkenazi Jewish families. Prenat Diagn. 2009;29:560 - 9. [PMC free article: PMC2735827] [PubMed: 19266496]
- Chijiiwa T, Nishimura M, Inomata H, Yamana T, Narazaki O, Kurokawa T. Ocular manifestations of congenital muscular dystrophy (Fukuyama type). Ann Ophthalmol. 1983;15:921 - 3, 926-8. [PubMed: 6651132]
- Cirak S, Foley AR, Herrmann R, Willer T, Yau S, Stevens E, Torelli S, Brodd L, Kamynina A, Vondracek P, Roper H, Longman C, Korinthenberg R, Marrosu G, Nürnberg P, Michele DE, Plagnol V, Hurles M, Moore SA, Sewry CA, Campbell KP, Voit T, Muntoni F, et al. ISPD gene mutations are a common cause of congenital and limb-girdle muscular dystrophies. Brain. 2013;136:269 - 81. [PMC free article: PMC3562076] [PubMed: 23288328]
- Devisme L, Bouchet C, Gonzalès M, Alanio E, Bazin A, Bessières B, Bigi N, Blanchet P, Bonneau D, Bonnières M, Bucourt M, Carles D, Clarisse B, Delahaye S, Fallet-Bianco C, Figarella-Branger D, Gaillard D, Gasser B, Delezoide AL, Guimiot F, Joubert M, Laurent N, Laquerrière A, Liprandi A, Loget P, Marcorelles P, Martinovic J, Menez F, Patrier S, Pelluard F, Perez MJ, Rouleau C, Triau S, Attié-Bitach T, Vuillaumier-Barrot S, Seta N, Encha-Razavi F. Cobblestone lissencephaly: neuropathological subtypes and correlations with genes of dystroglycanopathies. Brain. 2012;135:469 - 82. [PubMed: 22323514]
- Di Costanzo S, Balasubramanian A, Pond HL, Rozkalne A, Pantaleoni C, Saredi S, Gupta VA, Sunu CM, Yu TW, Kang PB, Salih MA, Mora M, Gussoni E, Walsh CA, Manzini MC. POMK mutations disrupt muscle development leading to a spectrum of neuromuscular presentations. Hum Mol Genet. 2014;23:5781 - 92. [PMC free article: PMC4189906] [PubMed: 24925318]
- Finsterer J, Ramaciotti C, Wang CH, Wahbi K, Rosenthal D, Duboc D, Melacini P. Cardiac findings in congenital muscular dystrophies. Pediatrics. 2010;126:538 - 45. [PubMed: 20679303]
- Fiorillo C, Moro F, Astrea G, Morales MA, Baldacci J, Marchese M, Scapolan S, Bruno C, Battini R, Santorelli FM. Novel mutations in the fukutin gene in a boy with asymptomatic hyperCKemia. Neuromuscul Disord. 2013;23:1010 - 5. [PubMed: 24144914]
- Fu X, Yang H, Jiao H, Wang S, Liu A, Li X, Xiao J, Yang Y, Wu X, Xiong H. Novel copy number variation of POMGNT1 associated with muscle-eye-brain disease detected by next-generation sequencing. Sci Rep. 2017;7:7056. [PMC free article: PMC5539251] [PubMed: 28765568]
- Gerin I, Ury B, Breloy I, Bouchet-Seraphin C, Bolsée J, Halbout M, Graff J, Vertommen D, Muccioli GG, Seta N, Cuisset JM, Dabaj I, Quijano-Roy S, Grahn A, Van Schaftingen E, Bommer GT. ISPD produces CDP-ribitol used by FKTN and FKRP to transfer ribitol phosphate onto α-dystroglycan. Nat Commun. 2016;7:11534. [PMC free article: PMC4873967] [PubMed: 27194101]
- Godfrey C, Clement E, Mein R, Brockington M, Smith J, Talim B, Straub V, Robb S, Quinlivan R, Feng L, Jimenez-Mallebrera C, Mercuri E, Manzur AY, Kinali M, Torelli S, Brown SC, Sewry CA, Bushby K, Topaloglu H, North K, Abbs S, Muntoni F. Refining genotype phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain. 2007;130:2725 - 35. [PubMed: 17878207]
- Godfrey C, Escolar D, Brockington M, Clement EM, Mein R, Jimenez-Mallebrera C, Torelli S, Feng L, Brown SC, Sewry CA, Rutherford M, Shapira Y, Abbs S, Muntoni F. Fukutin gene mutations in steroid-responsive limb girdle muscular dystrophy. Ann Neurol. 2006;60:603 - 10. [PubMed: 17044012]
- Godfrey C, Foley AR, Clemen E, Muntoni F. Dystroglycanopathies: coming into focus. Curr Opin Genet Dev. 2011;21:278 - 85. [PubMed: 21397493]
- Hayashi YK, Ogawa M, Tagawa K, Noguchi S, Ishihara T, Nonaka I, Arahata K. Selective deficiency of alpha-dystroglycan in Fukuyama-type congenital muscular dystrophy. Neurology. 2001;57:115 - 21. [PubMed: 11445638]
- Hill M, Hughes T, Milford C. Treatment for swallowing difficulties (dysphagia) in chronic muscle disease. Cochrane Database Syst Rev. 2004;2:CD004303. [PubMed: 15106246]
- Hino K, Fukuda M, Morino T, Ogata T, Ito M, Ishii E. Spinal fusion in a patient with Fukuyama congenital muscular dystrophy. Brain Dev. 2017;39:613 - 6. [PubMed: 28318781]
- Hino N, Kobayashi M, Shibata N, Yamamoto T, Saito K, Osawa M. Clinicopathological study on eyes from cases of Fukuyama type congenital muscular dystrophy. Brain Dev. 2001;23:97 - 107. [PubMed: 11248458]
- Kanagawa M, Kobayashi K, Tajiri M, Manya H, Kuga A, Yamaguchi Y, Akasaka-Manya K, Furukawa JI, Mizuno M, Kawakami H, Shinohara Y, Wada Y, Endo T, Toda T. Identification of a post-translational modification with ribitol-phosphate and its defect in muscular dystrophy. Cell Rep. 2016;14:2209 - 23. [PubMed: 26923585]
- Kanagawa M, Yu CC, Ito C, Fukada S, Hozoji-Inada M, Chiyo T, Kuga A, Matsuo M, Sato K, Yamaguchi M, Ito T, Ohtsuka Y, Katanosaka Y, Miyagoe-Suzuki Y, Naruse K, Kobayashi K, Okada T, Takeda S, Toda T. Impaired viability of muscle precursor cells in muscular dystrophy with glycosylation defects and amelioration of its severe phenotype by limited gene expression. Hum Mol Genet. 2013;22:3003 - 15. [PubMed: 23562821]
- Kang PB, Morrison L, Iannaccone ST, Graham RJ, Bönnemann CG, Rutkowski A, Hornyak J, Wang CH, North K, Oskoui M, Getchius TS, Cox JA, Hagen EE, Gronseth G, Griggs RC, et al. Evidence-based guideline summary: evaluation, diagnosis, and management of congenital muscular dystrophy: Report of the Guideline Development Subcommittee of the American Academy of Neurology and the Practice Issues Review Panel of the American Association of Neuromuscular & Electrodiagnostic Medicine. Neurology. 2015;84:1369 - 78. [PMC free article: PMC4388744] [PubMed: 25825463]
- Kato R, Kawamura J, Sugawara H, Niikawa N, Matsumoto N. A rapid diagnostic method for a retrotransposal insertional mutation into the FCMD gene in Japanese patients with Fukuyama congenital muscular dystrophy. Am J Med Genet A. 2004;127A:54 - 7. [PubMed: 15103718]
- Kato T, Funahashi M, Matsui A, Takashima S, Suzuki Y. MRI of disseminated developmental dysmyelination in Fukuyama type of CMD. Pediatr Neurol. 2000;23:385 - 8. [PubMed: 11118792]
- Kato Z, Morimoto M, Orii KE, Kato T, Kondo N. Developmental changes of radiological findings in Fukuyama-type congenital muscular dystrophy. Pediatr Radiol. 2010;40:S127 - 9. [PubMed: 20571791]
- Kato Z, Saito K, Isogai K, Kondo N. Magnetic resonance imaging and spectroscopy in Fukuyama-type congenital muscular dystrophy. J Pediatr Neurol. 2006;4:261 - 4.
- Kitamura Y, Kondo E, Urano M, Aoki R, Saito K. Target resequencing of neuromuscular disease-related genes using next-generation sequencing for patients with undiagnosed early-onset neuromuscular disorders. J Hum Genet. 2016;61:931 - 42. [PubMed: 27357428]
- Kobayashi K, Kato R, Kondo-Iida E, Taniguchi-Ikeda M, Osawa M, Saito K, Toda T. Deep-intronic variant of fukutin is the most prevalent point mutation of Fukuyama congenital muscular dystrophy in Japan. J Hum Genet. 2017;62:945 - 8. [PubMed: 28680109]
- Kobayashi K, Nakahori Y, Miyake M, Matsumura K, Kondo-Iida E, Nomura Y, Segawa M, Yoshioka M, Saito K, Osawa M, Hamano K, Sakakihara Y, Nonaka I, Nakagome Y, Kanazawa I, Nakamura Y, Tokunaga K, Toda T. An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature. 1998;394:388 - 92. [PubMed: 9690476]
- Kondo-Iida E, Kobayashi K, Watanabe M, Sasaki J, Kumagai T, Koide H, Saito K, Osawa M, Nakamura Y, Toda T. Novel mutations and genotype-phenotype relationships in 107 families with Fukuyama-type congenital muscular dystrophy (FCMD). Hum Mol Genet. 1999;8:2303 - 9. [PubMed: 10545611]
- Lim BC, Ki C-S, Kim J-W, Cho A, Kim MJ, Hwang H, Kim KJ, Hwang YS, Park WY, Lim Y-J, Kim IO, Lee JS, Chae JH. Fukutin mutations in congenital muscular dystrophies with defective glycosylation of dystroglycan in Korea. Neuromuscul Disord. 2010;20:524 - 30. [PubMed: 20620061]
- Lim BC, Lee S, Shin JY, Hwang H, Kim KJ, Hwang YS, Seo JS, Kim JI, Chae JH. Molecular diagnosis of congenital muscular dystrophies with defective glycosylation of alpha-dystroglycan using next-generation sequencing technology. Neuromuscul Disord. 2013;23:337 - 44. [PubMed: 23453855]
- Manzini MC, Gleason D, Chang BS, Hill RS, Barry BJ, Partlow JN, Poduri A, Currier S, Galvin-Parton P, Shapiro LR, Schmidt K, Davis JG, Basel-Vanagaite L, Seidahmed MZ, Salih MAM, Dobyns WB, Walsh CA. Ethnically diverse causes of Walker-Warburg syndrome (WWS): FCMD mutations are a more common cause of WWS outside of the Middle East. Hum Mutat. 2008;29:E231 - 41. [PMC free article: PMC2577713] [PubMed: 18752264]
- Manzini MC, Tambunan DE, Hill RS, Yu TW, Maynard TM, Heinzen EL, Shianna KV, Stevens CR, Partlow JN, Barry BJ, Rodriguez J, Gupta VA, Al-Qudah AK, Eyaid WM, Friedman JM, Salih MA, Clark R, Moroni I, Mora M, Beggs AH, Gabriel SB, Walsh CA. Exome sequencing and functional validation in zebrafish identify GTDC2 mutations as a cause of Walker-Warburg syndrome. Am J Hum Genet. 2012;91:541 - 7. [PMC free article: PMC3512000] [PubMed: 22958903]
- Meilleur KG, Zukosky K, Medne L, Fequiere P, Powell-Hamilton N, Winder TL, Alsaman A, El-Hattab AW, Dastgir J, Hu Y, Donkervoort S, Golden JA, Eagle R, Finkel R, Scavina M, Hood IC, Rorke-Adams LB, Bönnemann CG. Clinical, pathologic, and mutational spectrum of dystroglycanopathy caused by LARGE mutations. J Neuropathol Exp Neurol. 2014;73:425 - 41. [PMC free article: PMC5113964] [PubMed: 24709677]
- Mishima H, Hirata H, Ono H, Choshi K, Nishi Y, Fukuda K. A Fukuyama type of congenital muscular dystrophy associated with atypical gyrate atrophy of the choroid and retina. A case report. Acta Ophthalmol (Copenh). 1985;63:155 - 9. [PubMed: 4003043]
- Murakami T, Ishigaki K, Shirakawa S, Ikenaka H, Sakauchi M, Osawa M. Severe muscle damage following viral infection in patients with Fukuyama congenital muscular dystrophy. Brain Dev. 2012;34:293 - 7. [PubMed: 21726969]
- Nakanishi T, Sakauchi M, Kaneda Y, Tomimatsu H, Saito K, Nakazawa M, Osawa M. Cardiac involvement in Fukuyama-type congenital muscular dystrophy. Pediatrics. 2006;117:e1187 - 92. [PubMed: 16717122]
- Osawa M, Sumida S, Suzuki N, Arai Y, Ikenaka H, Murasugi H, Shishikura K, Suzuki H, Saito K, Fukuyama Y. Fukuyama type congenital muscular dystrophy. In: Fukuyama Y, Osawa M, Saito K, eds. Congenital Muscular Dystrophies. Amsterdam, Netherlands: Elsevier Science; 1997:31-68.
- Praissman JL, Willer T, Sheikh MO, Toi A, Chitayat D, Lin YY, Lee H, Stalnaker SH, Wang S, Prabhakar PK, Nelson SF, Stemple DL, Moore SA, Moremen KW, Campbell KP, Wells L. The functional O-mannose glycan on alpha-dystroglycan contains a phospho-ribitol primed for matriglycan addition. Elife. 2016:5. [PMC free article: PMC4924997] [PubMed: 27130732]
- Puckett RL, Moore SA, Winder TL, Willer T, Romansky SG, Covault KK, Campbell KP, Abdenur JE. Further evidence of Fukutin mutations as a cause of childhood onset limb-girdle muscular dystrophy without mental retardation. Neuromuscul Disord. 2009;19:352 - 6. [PMC free article: PMC2698593] [PubMed: 19342235]
- Riemersma M, Mandel H, van Beusekom E, Gazzoli I, Roscioli T, Eran A, Gershoni-Baruch R, Gershoni M, Pietrokovski S, Vissers LE, Lefeber DJ, Willemsen MA, Wevers RA, van Bokhoven H. Absence of α- and β-dystroglycan is associated with Walker-Warburg syndrome. Neurology. 2015;84:2177 - 82. [PubMed: 25934851]
- Saito K, Kobayashi M. Fukuyama congenital muscular dystrophy. In: Emery AEH, ed. Muscular Dystrophies. Oxford, UK: Oxford University Press; 2001:39-54.
- Saito W, Namba T, Inoue G, Imura T, Miyagi M, Nakazawa T, Shirasawa E, Uchida K, Takaso M. Spinal correction in patients with Fukuyama congenital muscular dystrophy. J Orthop Sci. 2017;22:658 - 64. [PubMed: 28325699]
- Saito Y, Mizuguchi M, Oka A, Takashima S. Fukutin protein is expressed in neurons of the normal developing human brain but is reduced in Fukuyama-type congenital muscular dystrophy brain. Ann Neurol. 2000;47:756 - 64. [PubMed: 10852541]
- Sato T, Murakami T, Ishiguro K, Shichiji M, Saito K, Osawa M, Nagata S, Ishigaki K. Respiratory management of patients with Fukuyama congenital muscular dystrophy. Brain Dev. 2016;38:324 - 30. [PubMed: 26363734]
- Signorino G, Covaceuszach S, Bozzi M, Hübner W, Mönkemöller V, Konarev PV, Cassetta A, Brancaccio A, Sciandra F. A dystroglycan mutation (p.Cys667Phe) associated to muscle-eye-brain disease with multicystic leucodystrophy results in ER-retention of the mutant protein. Hum Mutat. 2018;39:266 - 80. [PubMed: 29134705]
- Stevens E, Carss KJ, Cirak S, Foley AR, Torelli S, Willer T, Tambunan DE, Yau S, Brodd L, Sewry CA, Feng L, Haliloglu G, Orhan D, Dobyns WB, Enns GM, Manning M, Krause A, Salih MA, Walsh CA, Hurles M, Campbell KP, Manzini MC, Stemple D, Lin YY, Muntoni F, et al. Mutations in B3GALNT2 cause congenital muscular dystrophy and hypoglycosylation of alpha-dystroglycan. Am J Hum Genet. 2013;92:354 - 65. [PMC free article: PMC3591840] [PubMed: 23453667]
- Takaso M, Nakazawa T, Imura T, Okada T, Ueno M, Saito W, Takahashi K, Yamazaki M, Ohtori S. Surgical correction of spinal deformity in patients with congenital muscular dystrophy. J Orthop Sci. 2010;15:493 - 501. [PubMed: 20721717]
- Taniguchi K, Kobayashi K, Saito K, Yamanouchi H, Ohnuma A, Hayashi YK, Manya H, Jin DK, Lee M, Parano E, Falsaperla R, Pavone P, Van Coster R, Talim B, Steinbrecher A, Straub V, Nishino I, Topaloglu H, Voit T, Endo T, Toda T. Worldwide distribution and broader clinical spectrum of muscle-eye-brain disease. Hum Mol Genet. 2003;12:527 - 34. [PubMed: 12588800]
- Taniguchi M, Kurahashi H, Noguchi S, Sese J, Okinaga T, Tsukahara T, Guicheney P, Ozono K, Nishino I, Morishita S, Toda T. Expression profiling of muscles from Fukuyama-type congenital muscular dystrophy and laminin-alpha-2 deficient congenital muscular dystrophy; is congenital muscular dystrophy a primary fibrotic disease? Biochem Biophys Res Commun. 2006;342:489 - 502. [PubMed: 16487936]
- Taniguchi-Ikeda M, Kobayashi K, Kanagawa M, Yu C, Mori K, Oda T, Kuga A, Kurahashi H, Akman HO, DiMauro S, Kaji R, Yokota T, Takeda S, Toda T. Pathogenic exon-trapping by SVA retrotransposon and rescue in Fukuyama muscular dystrophy. Nature. 2011;478:127 - 31. [PMC free article: PMC3412178] [PubMed: 21979053]
- Taniguchi-Ikeda M, Morioka I, Iijima K, Toda T. Mechanistic aspects of the formation of α-dystroglycan and therapeutic research for the treatment of α-dystroglycanopathy: a review. Mol Aspects Med. 2016;51:115 - 24. [PubMed: 27421908]
- Tsutsumi A, Uchida Y, Osawa M, Fukuyama Y. Ocular findings in Fukuyama type congenital muscular dystrophy. Brain Dev. 1989;11:413 - 9. [PubMed: 2618965]
- Vannoy CH, Xiao W, Lu P, Xiao X, Lu QL. Efficacy of gene therapy is dependent on disease progression in dystrophic mice with mutations in the FKRP gene. Mol Ther Methods Clin Dev. 2017;5:31 - 42. [PMC free article: PMC5415313] [PubMed: 28480302]
- Voglmeir J, Kaloo S, Laurent N, Meloni MM, Bohlmann L, Wilson IB, Flitsch SL. Biochemical correlation of activity of the α-dystroglycan-modifying glycosyltransferase POMGnT1 with mutations in muscle-eye-brain disease. Biochem J. 2011;436:447 - 55. [PMC free article: PMC3133881] [PubMed: 21361872]
- Vuillaumier-Barrot S, Quijano-Roy S, Bouchet-Seraphin C, Maugenre S, Peudenier S, Van den Bergh P, Marcorelles P, Avila-Smirno D, Chelbi M, Romero NB, Carlier RY, Estournet B, Guicheney P, Seta N. Four Caucasian patients with mutations in the fukutin gene and variable clinical phenotype. Neuromusc Disord. 2009;19:182 - 8. [PubMed: 19179078]
- Wang CH, Bonnemann CG, Rutkowski A, Sejersen T, Bellini J, Battista V, Florence JM, Schara U, Schuler PM, Wahbi K, Aloysius A, Bash RO, Béroud C, Bertini E, Bushby K, Cohn RD, Connolly AM, Deconinck N, Desguerre I, Eagle M, Estournet-Mathiaud B, Ferreiro A, Fujak A, Goemans N, Iannaccone ST, Jouinot P, Main M, Melacini P, Mueller-Felber W, Muntoni F, Nelson LL, Rahbek J, Quijano-Roy S, Sewry C, Storhaug K, Simonds A, Tseng B, Vajsar J, Vianello A, Zeller R, et al. Consensus statement on standard of care for congenital muscular dystrophies. J Child Neurol. 2010;25:1559 - 81. [PMC free article: PMC5207780] [PubMed: 21078917]
- Watanabe M, Kobayashi K, Jin F, Park KS, Yamada T, Tokunaga K, Toda T. Founder SVA retrotransposal insertion in Fukuyama-type congenital muscular dystrophy and its origin in Japanese and Northeast Asian populations. Am J Med Genet A. 2005;138:344 - 8. [PubMed: 16222679]
- Xu L, Lu PJ, Wang CH, Keramaris E, Qiao C, Xiao B, Blake DJ, Xiao X, Lu QL. Adeno-associated virus 9 mediated FKRP gene therapy restores functional glycosylation of α-dystroglycan and improves muscle functions. Mol Ther. 2013;21:1832 - 40. [PMC free article: PMC3808132] [PubMed: 23817215]
- Yamamoto T, Taniguchi-Ikeda M, Awano H, Matsumoto M, Lee T, Harada R, Imanishi T, Hayashi N, Sakai Y, Morioka I, Takeshima Y, Iijima K, Saegusa J, Toda T. Cardiac involvement in Fukuyama muscular dystrophy is less severe than in Duchenne muscular dystrophy. Brain Dev. 2017;39:861 - 8. [PubMed: 28578814]
- Yang H, Kobayashi K, Wang S, Jiao H, Xiao J, Toda T, Wu X, Xiong H. Founder mutation causes classical Fukuyama congenital muscular dystrophy (FCMD) in Chinese patients. Brain Dev. 2015;37:880 - 6. [PubMed: 25814170]
- Yis U, Uyanik G, Heck PB, Smitka M, Nobel H, Ebinger F, Dirik E, Feng L, Kurul SH, Brocke K, Unalp A, Özer E, Cakmakci H, Sewry C, Cirak S, Muntoni F, Hehr U, Morris-Rosendahl DJ. Fukutin mutations in non-Japanese patients with congenital muscular dystrophy: less severe mutations predominate in patients with a non-Walker-Warburg phenotype. Neuromuscul Disord. 2011;21:20 - 30. [PubMed: 20961758]
- Yoshioka M. Phenotypic spectrum of fukutinopathy: most severe phenotype of fukutinopathy. Brain Dev. 2009;31:419 - 22. [PubMed: 18834683]
- Yoshioka M, Higuchi Y, Fujii T, Aiba H, Toda T. Seizure-genotype relationship in Fukuyama-type congenital muscular dystrophy. Brain Dev. 2008;30:59 - 67. [PubMed: 17597323]
- Yoshioka M, Kobayashi K, Toda T. Novel FKRP mutations in a Japanese MDC1C sibship clinically diagnosed with Fukuyama congenital muscular dystrophy. Brain Dev. 2017;39:869 - 72. [PubMed: 28629604]
本章节注解
更新历史
- 3 July 2019 (sw) 全面更新实时发布
- 10 May 2012 (me) 全面更新实时发布
- 26 January 2006 (me) 综述发布
- 8 October 2004 (ks) 初稿提交