
概述
临床特征.
Russell-Silver综合征(RSS) 是一类以胎儿宫内发育迟缓并伴随出生后生长缺陷为主要特点的综合征。典型的受累患儿出生时体重,身高低于平均值2个标准差及以下;受累个体常常表现为成比例的身材矮小,正常头围,小指弯斜,典型的脸部特征如具有突出前额的特征性三角形面容、窄下巴,身体受累侧生长迟缓引起偏侧肥大并进而导致的肢体不对称。RSS患儿的生长速度处于正常水平。RSS男性患者平均身高为151.2 cm,女性患者平均身高为139.9 cm。有证据表明,RSS患儿极有可能出现发育迟缓和学习障碍(包括运动和认知)。
诊断/检测.
RSS是一类遗传异质性较高的疾病。大多数受累个体具有有别于某一特定类型遗传病的表型。因此,诊断该病主要基于临床特征性表型的鉴定,尤其是胎儿宫内及出生后生长发育迟缓、正常头围。经鉴别,35%-50%的RSS患者在染色体11p15.5上的父源印迹中心1(IC1)处于低甲基化状态。 约10%的RSS患者的7号染色体为母源单亲二体性(mUPD7)。
临床管理.
对症治疗: 可考虑包括生长激素疗法; 身体、职业、说话和语言治疗; 以及个性化的教育计划。治疗胃食管反流,少食多餐和稠厚的饮食,推荐使用酸阻断药物(质子泵抑制剂或雷尼替丁)。 胃底折叠术等手术治疗可能仍有必要。 下肢长度超过3厘米的差异需要干预; 在大龄儿童中,可考虑骨延长术或者骺骨干固定术。 对于严重小颌畸形或腭裂的患儿,建议采用颅面部的多学科合作进行治疗。正颌手术不是必须的。男性隐睾症患者转诊至泌尿科; 根据需要决定是否进行睾丸固定术。男性小阴茎患者转诊至内分泌科; 根据需要决定是否进行雄激素激素治疗。
监控 :监测生长速度; 在有低血糖症状的婴儿和大龄儿童中监测血糖浓度; 早期儿童保健体检时监测肢体长度以提供肢体不对称增长的证据,并监测说话和语言能力的发展。
遗传咨询.
RSS病因存在多样化,包括:表观遗传学修饰的改变导致染色体11p15.5印迹区域基因表达的改变,母源性单亲二倍体(mUPD7),另外还有偶发的常染色体显性遗传、常染色体隐形遗传的模式。当一位患有RSS的先证者因父源性印记基因控制区1(IC1)发生低甲基化或母源性单亲二倍体(mUPD7)致病时,其父母双亲一般不受累,其同胞患此病的风险也不会超过正常人群,其后代患此病的风险通常很低。这是因为大多数RSS体现为单个家庭成员受累,大多数胎儿该病的发病风险并不确定会增加。因此,RSS的产前诊断通常不可行。经胎儿超声检查发现胎儿宫内发育迟缓的孕妇,可通过产前检查检测是否存在H19-IGF2父源性IC1区甲基化缺失和(或)7号染色体母源单亲二倍体(mUPD7)是可行手段。注:胎儿宫内发育迟缓通常在孕晚期才能确定。
诊断
临床诊断
RSS的临床诊断取决于是否有宫内发育迟缓并伴随出生后生长缺陷的临床表现[Silver et al 1953, Russell 1954, Price et al 1999]。诊断RSS没有其他标志性或特征性指征。尽管已经开发了一些针对RSS诊断的评分系统,Netchine et al [2007]和Bartholdi et al [2009]的研究主要集中在染色体11p15.5甲基化异常的个体的表型研究上(见 Testing). 此外,许多RSS患者缺乏典型的临床特征,具有不显著的临床表现,Eggermann et al [2009]的研究支持这一项观察性研究结果,该研究认为染色体11p15.5区发生表观遗传变异的RSS患者其临床表现通常更为和缓,如生长迟缓,身材不对称和前额突出等。Wakeling et al [2010]的研究也表明该病的临床特征在所有患者中并不都是一致的。根据Netchine et al [2007], Bartholdi et al [2009], Eggermann et al [2009]和 Wakeling et al [2010]的研究汇编的诊断标准(如下)指出:在进行RSS诊断以及进一步的实验室支持性检测时应该选择性考虑符合3个主要评价指标或2个主要评价指标附加2个次要评价指标的个体。
- 主要评价指标
- 宫内生长迟缓(低于同孕周胎儿10%)
- 出生后体重和身高增长速度低于平均值30%
- 正常头围(30%-97%)
- 四肢、身材和(或)面容不对称
- 次要评价指标
- 上肢臂短与正常的上臂-前臂比例
- 小指内弯
- 三角型面容
- 额头突出/突出前额
- 支持性评价指标
- 咖啡色斑或皮肤色素的变化
- 泌尿生殖系统异常(隐睾,尿道下裂)
- 运动,语言和(或)认知功能障碍
- 喂养困难
- 低血糖
Russell-Silver综合征(RSS) 是一类遗传异质性疾病(见 检测部分),具有一致性但差异化的表型:RSS患儿对生长激素的反应不同,不同程度的生长追赶延迟以及差异化的发育结局。
检测
已知的两类导致RSS的病因分别为染色体11p15.5上相关区域及7号染色体上相关区域。染色体11p15.5区相关的Russell-Silver综合征主要由于染色体11p15.5区印记的结构域异常所致[Abu-Amero et al 2010]。染色体11p15.5区的印记基因簇在胎儿和胎盘生长过程中发挥重要基因组印记。基因组印记是每个基因的等位基因DNA分子因差异化修饰导致仅来自亲本一方的单等位基因表达的现象;对于每个印记基因,所表达的亲本等位基因是特定的。印记基因经常在包含有调控作用的印记中心(IC)的基因簇中出现。 在染色体11p15.5区的其中一个印记基因簇中,亲本特异的差异性甲基化印记中心1(IC1)的调节IGF2(其编码对胎儿发育至关重要的生长因子)和H19(非编码转录本)的表达(图 1A)。 在RSS中,IC1的低甲基化导致H19双等位基因表达和IGF2双等位基因沉默,结果导致生长受限(图 1B)。参见 分子遗传学发病机制。
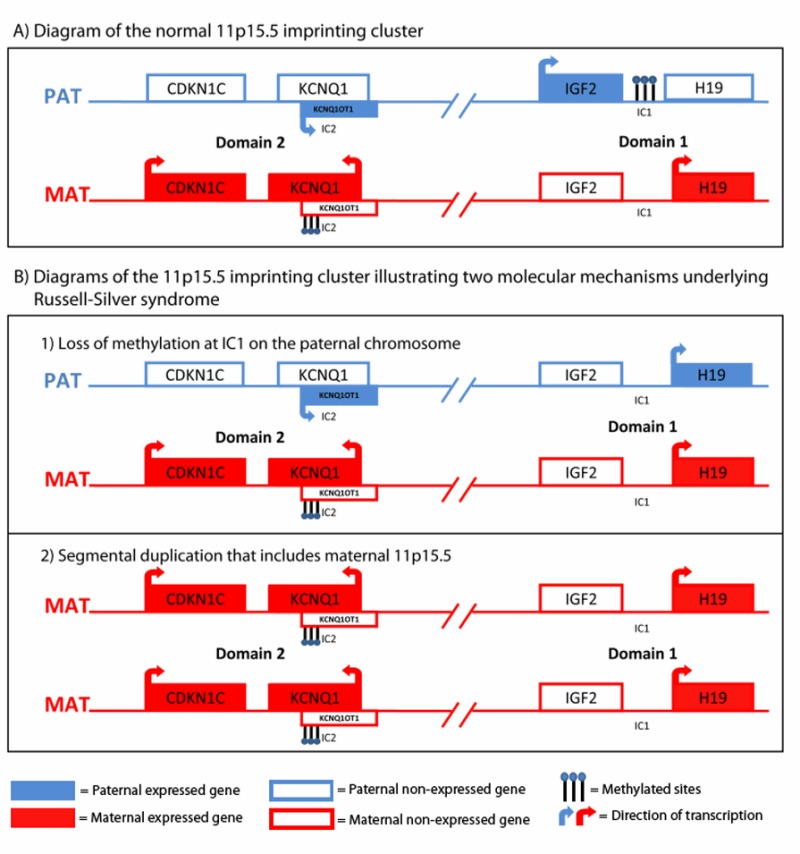
- 少数RSS患者仅H19 或 仅 IGF2 呈选择性低甲基化状态[Bartholdi et al 2009].
- 少数RSS患者其IGF2R (该基因编码染色体 6q25-q27上IGF2受体) 呈高甲基化状态,同时,其 H19 呈正常甲基化状态[Turner et al 2010].IGF2R的功能是清除循环的IGF2,从而限制其生长效应,在这种情况下,RSS可能因IGF2R的减少所致[Braulke 1999]。
- 少数RSS患者其母源性染色体11p15.5区存在重复。可通过细胞遗传的分析手段检测可能涉及易位和倒位的较大片段的重复 [Fisher et al 2002, Eggermann et al 2005], 但具有高分辨率的缺失/重复分析方法其敏感性更高 (Table 1)。RSS中的11p15.5的母源二倍体的发生机制尚不明确,其原因可能涉及CDKN1C上游印记的基因簇的剂量效应[Fisher et al 2002].
与7号染色体相关的RSS综合征
- 7号染色体母源单亲二体性(UPD7)约占RSS综合征病因的7%-10%[Moore et al 1999, Hannula et al 2001, Kim et al 2005]; 然而,导致UPD7印记的特定基因位置尚未确定(见 Molecular Genetic Pathogenesis)。
- 7号染色体母源部分单亲二体性已见报道:Hannula et al [2001] 报道了一例7号染色体母源部分单亲二体性(7q31-qter)的病例; Eggermann [2008]报道了两例7号染色体母源单亲二体性的病例,这两例均涉及大部分7号染色体长臂的单亲二体性(7q11.2-qter)。
- RSS个体中涉及7号染色体的罕见异常还包括以下情况:
- 在两例母源单亲二体性儿童中发现7号染色体三体型嵌合体[Flori et al 2005, Font-Montgomery et al 2005], 其中一例在产前得到鉴定[Font-Montgomery et al 2005]。
- 7号染色体p11.2-p12区段亚显微结构的重复经荧光原位杂交(FISH)得以鉴别 (常规核型分析未能识别,需要FISH或其他能鉴别缺失或重复的技术加以鉴定;见 Table 1) [Joyce et al 1999, Monk et al 2000]
见 Figure 1.
Table 1.
Russell-Silver Syndrome 分子遗传学检验
| RSS 类型 | 遗传学发病机制 | 检测方法 | 突变/改变的检测1 | 由该遗传机制引起的RSS的 比例 2 |
|---|---|---|---|---|
| 染色体 11p15.5 相关的 RSS | 染色体11p15.5区 IC1 甲基化 父源性缺失 | 甲基化分析 | IC1 父源性低甲基化水平 3, 4 | ~35%-50% |
| 染色体11p15.5区母源性重复 | 缺失/重复 分析 5 | 染色体11p15.5 区重复 | 未知 | |
| 7号染色体相关的 RSS | 单亲二体性UPD (母源性) | 单亲二体性UPD 分析 (多种方法) 6 | 7 号染色体母源单亲二体性 7 | ~7%-10% |
| 缺失/ 重复 | 缺失/重复 分析, 细胞遗传的 分析方法 | 7号染色体异常 | 罕见 |
2.检测方法的选择常常是基于遗传机制和染色体位置的改变
3.嵌合体可能导致假阴性结果,这可能由于染色体11p15.5区低甲基化发生在受精卵形成后。可对来自其他组织的样本进行再次检测(例如口腔细胞,成纤维细胞等)。
4.染色体11p15.5区特异性甲基化检测不推荐用于产前诊断, 这是因为在胚胎期,特定位置甲基化的时期不确定。
5.区别外显子或多个基因缺失或重复的检测方法,通过对基因组的DNA编码区和内含子的侧翼区域的序列分析并不容易检测到这些变异 ; 这些方法包括可以使用的各种检测手段:定量 PCR,长片段PCR,多重连接探针扩增(MLPA)和包含该基因/染色体片段的染色体芯片 (CMA)。
6.UPD的检测有多种方法,例如:SNP或分子标记分析,MS-MLPA(甲基化特异性多重连接探针扩增)。 检测可能需要亲本的血液样本。
7.已经在7号染色体 母源单亲二体性以及其他7号染色体重排的病例中观察到嵌合体,检测其他来源的组织样本可能更为合适。
临床特征
临床表现
用于诊断的关键性临床特征[Price et al 1999]:
- 宫内发育迟缓(IUGR): 出生时体重低于平均值2个标准差(SD)或更多。
- 出生后发育迟缓: 长度或身高低于平均值2个标准差(SD)或更多。
- 头围正常,常伴随“假性脑积水”。
- 小指弯曲
- 肢体长度不对称
可辅助诊断的其他临床特征:
- 身材矮小,上下肢比例正常,骨骼检查正常,经常性的骨龄延迟
- 典型的面部表型:宽且突出的额头,小三角脸,窄小的下巴,下翻的嘴角
- 低血糖症
- 短指,屈曲指
- 咖啡斑
- 手臂跨度低于高度
生长 RSS儿童的早期问题通常与生长和喂养有关。 RSS患儿宫内发育迟缓,且出生后生长缺陷。欧洲RSS患儿的生长参数图表已经发表[Wollmann et al 1995]. 北美RSS患儿的生长图表可以从MAGIC 基金会获得。
生长速度正常。在未接受生长激素治疗的RSS患者中,成年男性平均身高为151.2 cm(-7.8 SD),成年女性平均身高为139.9 cm(-9 SD)[Wollmann et al 1995]。预计可成比例生长,尽管大多数RSS患者的手臂跨度与身高相比较短,而身材上,下段比例正常[Silver et al 1953, Saal et al 1985]。参见 Management ,使用生长激素疗法来干预RSS患儿的生长。注意:即使采用人生长激素进行管控,许多RSS患儿也没有达到正常的身材。
大多数据说患有RSS的孩子在童年后期表现出追赶式的成长[Saal et al 1985] ,这些孩子的情况可能不同于经典的。
生长激素缺乏 一项对24名RSS患儿的研究发现:这些研究对象中,有10名儿童发生低血糖;在几名儿童中被发现生长激素不足(胰高血糖素刺激试验确定),并被认为是低血糖的一个可能原因[Azcona & Stanhope 2005]。
骨骼畸形 RSS个体通常仅仅是肢体长度不对称,至少在某些个体中可能是受累的一侧生长减少导致的单侧生长不足。骨骼畸形作为RSS的诊断标准之一,在RSS患者中,小指弯曲是最常见的骨骼畸形之一。一项系统性的研究发现25名有骨骼畸形的RSS患者中,19名存在掌骨和趾骨异常,9名脊柱侧凸,5名脚趾并指,3名髋关节发育不良[Abraham et al 2004]。
神经发育 除了生长方面的问题之外,神经发育可能是RSS患者父母最关心的问题。 尽管在早期病例报道中, RSS患者“智力正常”得到反复确认,但越来越多的证据表明,RSS患儿在发育迟缓(运动和认知)和学习障碍方面存在显著风险。
- 在一项针对20名6至12岁RSS患儿的研究中,平均智商为86。此外,这些儿童中有36%需要接受特殊教育,48%需要语言方面的治疗[Lai et al 1994]。该项研究中对于入组患儿并未确定RSS的具体病因。
- 在另一项研究中,36名RSS患儿的平均智商为95.7,其同胞对照组为104.20。 值得注意的是,两个7号染色体母源单亲二体性致病的患儿其智商分别为81和84 [Noeker & Wollmann 2004]。
- 在一项因染色体11p15甲基化 缺陷或母源UPD7致病的RSS患者的大型队列回顾性研究中,轻微发育迟缓在母源UPD7致病的患者中(65%)比11p15甲基化缺陷致病的患者中(20%)更为常见。语言发育迟缓在两组中均常见[Wakeling et al 2010]。
低血糖症 RSS患儿几乎没有皮下脂肪,很瘦,而且往往食欲不振; 如较长时间未摄取食物,包括手术中禁食,这些患儿有低血糖发生风险[Tomiyam a et al 1999]。在一项针对RSS患儿的研究中,影响低血糖发生的因素包括热量摄入减少,这通常在食欲不振和不良喂食后继发; 体重减轻; 以及发生在少数几个患儿中的生长激素缺乏症[Azcona & Stanhope 2005]。虽然大多数RSS患儿有低血糖的临床症状,特别是出汗(过度出汗),其中一些仍为无低血糖症状患儿。
儿童早期的发汗可能与低血糖相关,尽管没有发生低血糖时也可能会发汗[Stanhope et al 1998]。
胃肠道疾病 较为常见[Anderson et al 2002]。这些疾病包括包括胃食管反流、食管炎,厌食和发育停滞。 其中一些问题可能是医源性的(例如,与生长缓慢治疗有关的)。患有厌食或误吸的儿童应怀疑反流性食管炎。
严重的颅面畸形 并不常见。一些RSS个体拥有Pierre Robin综合征和腭裂。Wakeling et al [2010] 发现7%的染色体11p15.5found 甲基化缺陷患者中发现患有腭裂或悬雍垂裂,而在母源性UPD7患者中未发现此类症状。
牙齿和口腔畸形很少见。小牙畸形,高拱形腭以及因相对小颌畸形和小口继发的牙齿拥挤也已有报道[Cullen & Wesley 1987, Kulkarni et al 1995, Orbak et al 2005, Wakeling et al 2010]。在牙齿拥挤的情况下,口腔卫生不佳可能导致龋齿的风险增加。
泌尿生殖系统的问题 已经有观察报道,但并不常见。 最常见的异常是尿道下裂和隐睾。 已报道的肾脏方面的异常,包括肾盂积水,肾小管酸中毒,后尿道瓣膜症和马蹄肾[Arai et al 1988, Ortiz et al 1991]。
肿瘤 尽管偶尔有RSS个体罹患恶性肿瘤的报道,包括Wilms瘤,肝细胞肝癌和颅咽管瘤,但RSS个体的肿瘤发病率似乎并未显著增加[Draznin et al 1980, Chitayat et al 1988, Bruckheimer & Abrahamov 1993].
基因型-表型的相关性
Bruce et al [2009] 利用甲基化敏感的限制性内切酶HpaII或NotI来评估H19的甲基化程度,并发现了不同程度的H19低甲基化水平,包括极低、中度、正常水平和母源性UPD7(H19甲基化正常水平)。他们确定,存在H19极低甲基化水平(即≤-6SD或<9%甲基化程度)的RSS患儿比中度H19低甲基化水平的RSS患儿以及因母源性UPD7致病的RSS患儿更可能表现出更为严重的骨骼表型(包括桡骨错位,并指/趾,更大程度的肢体不对称性和脊柱侧凸)。
Wakeling et al [2010] 的一项研究比较了IC1甲基化缺陷致病和母源性UPD7致病的RSS患儿的临床特征。 他们发现所致表型存在相当大的重叠:在IC1甲基化缺陷致病的RSS患儿中,小指弯曲和先天畸形发生率比因母源性UPD7致病的RSS患儿更常见,而因母源性UPD7致病的RSS患儿的学习和语言障碍比因IC1甲基化缺陷致病的RSS患儿更常见。
RSS个体患恶性肿瘤的风险低 ,鉴于有一部分RSS个体在染色体11p15.5的印记的区域发生了突变,发生Wilms瘤、肝母细胞瘤及其他腹部肿瘤而后者与Beckwith-Wiedemann综合征患者有关, 肿瘤发病风险似乎随着与过度生长相关的突变而增加,而与发育迟缓相关的突变而减少。
流行病学
RSS的发病率据评估约为1/ 100,000 [Christoforidis et al 2005]。
相关(等位基因的)的遗传病
Beckwith-Wiedemann 综合征与染色体11p15.5(也称为BWS关键区域)印记的结构域中基因转录的异常调节有关。
已有报道,在孤立的单侧发育过度的个体中11p15区域的分子生物学改变,包括IC2甲基化丢失,IC1甲基化增加[Martin et al 2005]和11p15父源单亲二体性 [Shuman et al 2002] 。
父源的IC1甲基化丢失的体细胞嵌合与孤立的单侧发育迟缓有关[Zeschnigk et al 2008, Eggermann 2009]。
单纯的Wilms 瘤 可与染色体11p15.5区域的组成性改变相关,包括IC1的高甲基化,11p15.5父源单亲二体性以及基因组异常(包括微缺失和微插入) [Scott et al 2008]。
鉴别诊断
宫内发育迟缓与身材矮小。RSS综合征的鉴别诊断包括任何可能导致宫内发育迟缓和身材矮小的病症。
染色体异常和缺失/重复的分析。许多由染色体失衡导致的疾病可能被误诊为RSS综合征,由于很多因染色体不平衡导致的疾病可能被误诊为RSS,发现有RSS类似症状的儿童应该进行染色体分析(最好进行高分辨率染色体芯片分析,包括比较基因组杂交和SNP微阵列芯片),其检出率高于常规的细胞遗传的分析。
在RSS的鉴别诊断中要考虑的染色体异常包括:
- Yq 缺失 [Leppig et al 1991]
- 二倍体/三倍体混倍性(由于存在肢体不对称) [Graham et al 1981]
- 嵌合型Turner综合征 [Li et al 2004]
- 患有小头畸形和智力障碍的个体中存在12p14缺失,同时也有一些Russell-Silver综合征的提示性特征[Spengler et al 2010]。
- 15q26.3缺失(包括IGF1R)和22q11.2的远端缺失(已知该区域与宫内发育迟缓相关)[Bruce et al 2009]。
- 17q25重排 [Ramírez-Dueñas et al 1992, Midro et al 1993]。
DNA 修复异常,包括Fanconi 贫血, Nijmegen 断裂综合征,和Bloom 综合征在内的疾病常常与宫内发育迟缓和身材矮小有关。在这些情况下,其他附加的临床特征,包括小头畸形,阳光敏感性皮肤和肢体异常通常较为明显。
其他
- 一直以来,与RSS混淆的一个疾病是 X染色体连锁 的身材矮小与皮肤色素沉着过度相关的疾病。Partington [1986] 描述了第一个病例,并将其称为X染色体连锁的RSS综合征。 在没有阳性家族史的情况下,这种疾病可能难以与经典RSS综合征区分开来。
- 胎儿酒精综合征(FAS)患儿通常发生胎儿宫内发育迟缓,小头畸形,发育停滞,并且通常为三角脸型。 大多数FAS患儿出生前的乙醇暴露可有记录在案,并且其面部表型(短睑裂,扁平人和薄上唇)往往有特征性。
- IMAGe 综合征 的特征是宫内生长受限,干骺端发育不良,先天性肾上腺发育不全,以及包括隐睾和小阴茎在内的生殖器畸形。头围正常[Vilain et al 1999, Pedreira et al 2004。IMAGe综合征是由母系传递的CDKN1C致病性变异 所导致。
- 应进行骨骼检查以排除可能雷同于RSS的骨骼发育不良症状。
注意:RSS患儿的骨龄可能延迟; 然而,在由多种病因导致的宫内发育迟缓患儿中,骨龄延迟并非特征性表现。
小头畸形 RSS个体的头围正常。当头围低于平均值3个SD以上时,应该考虑其他导致发育迟缓的病因。
管理
初诊后评估
为了确定患有RSS综合征个体的疾病严重程度,建议进行以下评估:
- 估并绘制生长曲线。对于欧洲的儿童,请参阅Wollmann et al [1995]; 对于北美的儿童,请参阅 MAGIC Foundation。
- 体格检查用于评估可能的肢体大小的不对称程度和口腔、颅面异常
- 对于大多数RSS患儿,采用标准法评估生长激素缺乏症
- 对于出现发汗的儿童,评估低血糖
- 对于怀疑患有胃食管反流(GERD)的儿童,进行食管炎的评估,采用包括钡剂吞咽研究,pH探头和内镜检查等技术方法
- 神经认知发展、语言和肌肉张力的筛检评估
对症治疗
生长 患有身体方面异常和/或身材矮小的儿童往往对自身敏感。这些可能在自我形象形成,同伴关系和社会化等方面有重要影响。 因此,心理咨询通常对患有RSS的儿童有帮助。
人生长激素在治疗因各种病因导致的宫内发育迟缓儿童可以显著改善生长和最终身高[Albanese & Stanhope 1997, Azcona et al 1998, Czernichow & Fjellestad-Paulsen 1998, Saenger 2002]。具体而言,即使没有出现生长激素缺乏症的RSS儿童[Albanese & Stanhope 1997],也能在补充生长激素的治疗中获益,包括显著的生长加速和最终身高的增加[Azcona et al 1998] 以及终止治疗后的持续的正常生长速度[Azcona & Stanhope 1999]。这种治疗最好在具有治疗生长障碍经验的中心进行。一项研究表明,用生长激素治疗的RSS患儿身高显著增加,但身体或肢体的不对称性没有改变[Rizzo et al 2001]。与11p15.5表观遗传突变的个体相比,具有UPD7的RSS患儿在采用生长激素治疗时身高增幅更高,这可能是因为11p15.5甲基化异常的RSS患儿表现出胰岛素样生长因子II(IGF2的产物)水平升高; 具有UPD7的RSS患儿的反应特征与其他小于胎龄的儿童相似[Binder et al 2008]。一项关于IGF1和IGF结合蛋白-3(IGFBP-3)水平的后期研究显示,采用生长激素治疗后,这些蛋白水平的变化与生长速度之间没有相关性; 然而,本研究中RSS的诊断仅基于临床表现,并无染色体11p15区甲基化缺陷或母源UPD7的相关检测资料。
[Beserra et al 2010]。在一项最近的长期结果研究中,采用生长激素治疗有显著相应:26名RSS患儿接受生长激素治疗的时间中位数为9.8年,治疗开始时为中位数身高-2.7标准差,治疗结束时为中位身高-1.3SD[Toumba et al 2010]。遗憾的是,鉴于已知的疾病的遗传异质性以及许多研究中病因学资料的缺乏,很难说明这些研究中接受生长激素治疗的RSS患儿的治疗结果。同样的,研究生长激素治疗对RSS患儿的长期影响是十分重要的,尤其是该疗法对于最终成年身高的影响以及肢体长度不对称的患病个体的矫形管理可能带来的改变。
生长激素缺乏症 在存在生长激素缺乏症的情况下,用人类生长激素治疗是必要的。
骨骼异常 由于下肢长度超过3厘米的差异可导致代偿性脊柱侧弯,因此需要干预。初步的处理是使用增高鞋垫。在大龄儿童中,可以考虑骨延长术或者骺骨干固定术。
神经发育
- 对于肌张力低下的婴儿,请转诊至早期干预项目和理疗师
- 对于有发育延迟证据的儿童,请早期干预并进行言语和语言治疗
- 对于学龄儿童,通过进行适当的神经心理学测试和制订个性化教育计划,并与学校办学模式结合起来解决学习障碍的问题
低血糖症应通过标准化方式进行治疗,包括膳食补充,频繁喂食和使用复合碳水化合物。
胃肠道疾病应积极管理。
- 治疗胃食管反流推荐首先进行定位和增稠喂食,同时使用酸阻断药物(最好是质子泵抑制剂,如奥美拉唑或帕托普拉唑)。在较为严重的情况下或采用保守治疗未果,可能需要行胃底折叠术等手术治疗。
- 厌食反应可通过言语病理学家和/或职业治疗师的治疗来解决。
颅面异常 对于严重小颌畸形或腭裂的患儿,建议采用颅面部的多学科合作进行治疗。正颌手术很少是必须的。
口腔卫生和牙列拥挤可以由儿科牙医和正牙医生按常规方式适当管理。泌尿生殖系统异常
- 转诊男性隐睾症患者至泌尿科; 根据需要决定是否手术
- 转诊男性小阴茎患者至内分泌科; 根据需要决定是否进行雄激素激素治疗
肿瘤 RSS患者的恶性肿瘤风险较低。虽然可能存在身体不对称,但它似乎并没有Beckwith-Wiedemann 综合征所表现的偏侧肥大;因此,对于RSS患儿,不推荐常规进行连续的经腹和肾脏超声。
监控
适当的做法如下:
- 监测生长并特别关注生长速度
- 在患有发汗或食欲不振的婴儿期和大龄儿童中,监测血糖浓度以管理低血糖
- 在幼年早期进行体检,检查并测量肢体长度差异
- 密切监控说话和语言发育情况
高危亲属的风险评估
请参阅 Genetic Counseling所列有关高危亲属为遗传咨询遗传咨询 进行检测的问题。
正在进行研究的疗法
请至 ClinicalTrials.gov 获取关于各种疾病的临床研究信息。 注意:该疾病可能没有相关临床试验。
遗传咨询
遗传咨询是向个人和家庭提供有关遗传性疾病的性质、遗传方式以及影响等信息以帮助他们在充分知情的情况下做出医疗和个人决定的过程。 以下部分为明确家庭成员的遗传信息涉及的遗传风险评估以及家族史、基因检测的应用。 本部分不是要解决个人可能面临的所有个性化的、文化背景或伦理的争议,也不能替代与遗传专业人士的咨询。 —编者.
遗传模式
RSS综合征的病因有多种,包括父源H19-IGF2的印迹中心1(IC1)甲基化丢失和母源性7号染色体单亲二体性(UPD7) (见 Testing)。 常染色体显性遗传或常染色体隐性遗传方式比较罕见[Ounap et al 2004]。
家族成员面临的风险 - 染色体11p15.5区域相关的RSS:父源H19-IGF2 IC1上甲基化丢失
先证者父母
- Bartholdi et al [2009]报道了一个父亲和女儿共同患有RSS的案例,父女二人在H19-IGF2 IC1处均存在甲基化缺陷。
- 表观遗传突变在子代中得以鉴定,由此推断两个家族中未受累的父亲中存在生殖嵌合[Bartholdi et al 2009]。
先证者的同胞
先证者的子代
家庭成员的风险 - 与7号染色体相关的RSS:7号染色体母源单亲二体性
先证者父母 先证者为母源单亲二体性(UPD7)导致的RSS时,父母双方预计均不受累。
先证者的同胞 先证者为母源单亲二体性(UPD7)导致的RSS时,同胞的风险不会高于正常人群。
相关的遗传咨询问题
经验风险 45%-60%经鉴定存在11p15区域低甲基化或母源单亲二体性(UPD7)的个体中,其父母和同胞身材正常,同胞的复发风险并未增加,而是与正常人群相同。
DNA银行是指DNA存储(通常由白细胞提取)以备用。 由于检测方法学和我们对基因、等位基因变异体和疾病的理解可能进一步加深,受累的个体应给予DNA备份的考虑建议。
产前检查
由于RSS的发生大部分只涉及单个家庭成员,大多数妊娠的患病风险未被确定为增加。因此,RSS的产前诊断通常是不可行的。
对于经胎儿超声检查发现存在宫内发育迟缓的孕妇,通过PCR分子检测技术来检测经羊膜穿刺术获得的亲本和胎儿细胞,进行产前检测H19-IGF2 IC1区父源性甲基化丢失和母源性UPD7是可行的。注:胎儿宫内发育迟缓通常在孕晚期才能完全确定。
资源
GeneReviews工作人员选择了以下基于该疾病特定的/或支持组织和/或注册中心,以帮助那些患有该种疾病的个体和他们的家庭。GeneReviews对其他组织提供的信息不负有相关责任。有关选择标准的信息,请点击here。
- MAGIC Foundation6645 West North AvenueOak Park IL 60302Phone: 708-383-0808Fax: 708-383-0899Email: ContactUs@magicfoundation.org
- Silver-Russell Support Groupc/o Child Growth Foundation2 Mayfield AvenueChiswick WA 1PWUnited KingdomPhone: 020 8995 0257; 020 8994 7625Fax: 020 8995 9075
- Child Growth FoundationUnited Kingdom
- Human Growth Foundation (HGF)997 Glen Cove AvenueSuite 5Glen Head NY 11545Phone: 800-451-6434 (toll-free)Fax: 516-671-4055Email: hgf1@hgfound.org
- Little People of America, Inc. (LPA)250 El Camino RealSuite 201Tustin CA 92780Phone: 888-572-2001 (toll-free); 714-368-3689Fax: 714-368-3367Email: info@lpaonline.org
分子遗传学
分子遗传学和OMIM的表格中的信息可能与GeneReview中其他地方提供的信息不同:这些表格可能包含更多的最新信息。-编者.
表 A.
Russell-Silver 综合征: 基因和数据库
| 基因 | 染色体位置 | 蛋白 | Locus-Specific 数据库 | HGMD | ClinVar |
|---|---|---|---|---|---|
| H19 | 11p15-.5 | 未知 | H19 @ LOVD | H19 | H19 |
| IGF2 | 11p15-.5 | Insulin-like growth factor II | LOVD - Growth Consortium (IGF2) | IGF2 | IGF2 |
| 未知 | 7号染色体 | 未知 |
表 B.
Russell-Silver 综合征的OMIM 入口(View All in OMIM)
分子遗传学发病机制
染色体11p15.5区域相关的RSS 已知染色体11p15.5上的印记基因对于胎儿生长是十分重要的[DeChiara et al 1990, Fitzpatrick et al 2002, Eggermann 2009]。RSS因印记结构域1的表观遗传改变所导致[Gicquel et al 2005, 图 1],而表现为过度生长障碍的Beckwith-Wiedemann 综合征由印记区域1和2的表观遗传改变导致 (关于两者的对比,请参见 Beckwith-Wiedemann 综合征, 图 1)。11p15.5上的差异甲基化区域影响基因转录的机制尚不明确。一个模型指出,印记中心1(IC1)结合锌指CTCF蛋白可以控制染色质构象,进而导致染色质结构域的激活或失活[Li et al 2008, Demars et al 2010]。对于RSS,结构域1低甲基化诱导的染色质结构改变阻断了来自顺式作用原件--增强子序列和/或调控蛋白的转录信号,进而导致IGF2关闭并允许H19双等位基因的表达。与H19-IGF2和IC1相关的RSS的研究包括 Obermann et al [2004], Gicquel et al [2005], Schönherr et al [2007]和Turner et al [2010]。
- IGF2 是一个父源化印记的 表达转录本,编码的胰岛素家族成员是一类多肽生长因子,参与发育和生长。NM_000612.4, 是最主要的转录本,编码180个氨基酸残基,为胰岛素样生长因子II异构体 (NP_000603.1)。
7号染色体相关的RSS 导致UPD7印记的具体基因座尚未确定; 然而,考虑到7号染色体长臂(7q)的母源性UPD的病例报道,感兴趣的基因可能位于7号染色体长臂(7q)[Eggermann 2008]。Hannula et al [2001] 报道了一例7q31-qter因母源性UPD致病的患者。
参考文献
已发布的指南/专家共识
- American College of Medical Genetics Statement on diagnostic testing for uniparental disomy. Available online. 2001. Accessed 3-17-16.
引用的文献
- Abraham E, Altiok H, Lubicky JP. Musculoskeletal manifestations of Russell-Silver syndrome. J Pediatr Orthop. 2004;24:552-64. [PubMed: 15308907]
- Abu-Amero S, Wakeling EL, Preece M, Whittaker J, Stanier P, Moore GE. Epigenetic signatures of Silver-Russell syndrome. J Med Genet. 2010;47:150-54. [PubMed: 20305090]
- Albanese A, Stanhope R. GH treatment induces sustained catch-up growth in children with intrauterine growth retardation: 7-year results. Horm Res. 1997;48:173-7. [PubMed: 9378463]
- Anderson J, Viskochil D, O'Gorman M, Gonzales C. Gastrointestinal complications of Russell-Silver syndrome: a pilot study. Am J Med Genet. 2002;113:15-9. [PubMed: 12400060]
- Arai Y, Wakabayashi Y, Pak K, Tomoyoshi T. Horseshoe kidney in Russell-Silver syndrome. Urology. 1988;31:321-3. [PubMed: 2895527]
- Azcona C, Albanese A, Bareille P, Stanhope R. Growth hormone treatment in growth hormone-sufficient and -insufficient children with intrauterine growth retardation/Russell-Silver syndrome. Horm Res. 1998;50:22-7. [PubMed: 9691209]
- Azcona C, Stanhope R. Absence of catch-down growth in Russell-Silver syndrome after short-term growth hormone treatment. Horm Res. 1999;51:47-9. [PubMed: 10095170]
- Azcona C, Stanhope R. Hypoglycaemia and Russell-Silver syndrome. J Pediatr Endocrinol Metab. 2005;18:663-70. [PubMed: 16128243]
- Bartholdi D, Krajewska-Walasek M, Ounap K, Gaspar H, Chrzanowska KH, Ilyana H, Kayserili H, Lurie IW, Schinzel A, Baumer A. Epigenetic mutations of the imprinted IGF2-H19 domain in Silver-Russell syndrome (SRS): results from a large cohort of patients with SRS and SRS-like phenotypes. J Med Genet. 2009;46:192-7. [PubMed: 19066168]
- Bernard LE, Penaherrera MS, Van Allen MI, Wang MS, Yong SL, Gareis F, Langlois S, Robinson WP. Clinical and molecular findings in two patients with Russell-Silver syndrome and UPD7: comparison with non-UPD7 cases. Am J Med Genet. 1999;87:230-6. [PubMed: 10564876]
- Beserra IC, Ribeiro MG, Collett-Solberg PF, Vaisman M, Guimarães MM. IGF-I and IGF binding protein-3 generation tests and response to growth hormone in children with Silver-Russell syndrome. Int J Pediatr Endocrinol. 2010;2010:546854. [PMC free article: PMC3017907] [PubMed: 21234390]
- Binder G, Seidel AK, Martin DD, Schweizer R, Schwarze CP, Wollmann HA, Eggermann T, Ranke MB. The endocrine phenotyupe in Silver-Russell syndrome is defined by the underlying epigenetic alteration. J Clin Endocrinol Metab. 2008;93:1402-7. [PubMed: 18230663]
- Braulke T. Type-2 IGF receptor: a multi-ligand binding protein. Horm Metab Res. 1999;31:242-6. [PubMed: 10226808]
- Bruce S, Hannula-Jouppi K, Peltonen J, Kere J, Lipsanen-Nyman M. Clinically distinct epigenetic subgroups in Silver-Russell syndrome: the degree of H19 hypomethyulation associates with phenotype severity and genital and skeletal anomalies. J Clin Endocrinol Metab. 2009;94:579-87. [PubMed: 19017756]
- Bruckheimer E, Abrahamov A. Russell-Silver syndrome and Wilms tumor. J Pediatr. 1993;122:165-6. [PubMed: 8380450]
- Chitayat D, Friedman JM, Anderson L, Dimmick JE. Hepatocellular carcinoma in a child with familial Russell-Silver syndrome. Am J Med Genet. 1988;31:909-14. [PubMed: 2853572]
- Christoforidis A, Maniadaki I, Stanhope R. Managing children with Russell-Silver syndrome: more than just growth hormone treatment? J Pediatr Endocrinol Metab. 2005;18:651-2. [PubMed: 16128241]
- Courtens W, Vermeulen S, Wuyts W, Messiaen L, Wauters J, Nuytinck L, Peeters N, Storm K, Speleman F, Nöthen MM. An interstitial deletion of chromosome 7 at band q21: a case report and review. Am J Med Genet A. 2005;134A:12-23. [PubMed: 15732063]
- Cullen CL, Wesley RK. Russell-Silver syndrome: microdontia and other pertinent oral findings. ASDC J Dent Child. 1987;54:201-4. [PubMed: 3473100]
- Czernichow P, Fjellestad-Paulsen A. Growth hormone in the treatment of short stature in young children with intrauterine growth retardation. Horm Res. 1998;49 Suppl 2:23-7. [PubMed: 9716823]
- DeChiara TM, Efstratiadis A, Robertson EJ. A growth-deficiency phenotype in heterozygous mice carrying an insulin-like growth factor II gene disrupted by targeting. Nature. 1990;345:78-80. [PubMed: 2330056]
- Demars J, Ennuri Shmela M, Rossignol S, Okabe J, Netchine I, Azzi S, Cabrol S, Le Caignec C, David A, Le Bouc Y, El-Osta A, Cicquel C. Analysis of the IFG2/H19 imprinting control region uncovers new genetic defects, including mutations of OCT-binding sequences in patients with 11p15 fetal growth disorders. Hum Mol Genet. 2010;19:803-14. [PubMed: 20007505]
- Draznin MB, Stelling MW, Johanson AJ. Silver-Russell syndrome and craniopharyngioma. J Pediatr. 1980;96:887-9. [PubMed: 7189211]
- Eggermann T. Segmental maternal UPD(7q) in Silver-Russell syndrome. Clin Genet. 2008;74:486-9. [PubMed: 18700897]
- Eggermann T. Silver-Russell and Beckwith-Wiedemann syndromes: opposite (epi)mutations in 11p15 result in opposite clinical pictures. Horm Res. 2009;71 Suppl 2:30-5. [PubMed: 19407494]
- Eggermann T, Begemann M, Binder G, Spengler S. Silver-Russell syndrome: genetic basis and molecular genetic testing. Orphanet J Rare Dis. 2010;5:19. [PMC free article: PMC2907323] [PubMed: 20573229]
- Eggermann T, Gonzalez D, Spengler S, Arslan-Kirchner M, Binder G, Schonherr N. Broad clinical specrumin Silver-Russell syndrome and consequences for genetic testing in growth retardation. Pediatrics. 2009;123:e929-31. [PubMed: 19364767]
- Eggermann T, Meyer E, Obermann C, Heil I, Schüler H, Ranke MB, Eggermann K, Wollmann HA. Is maternal duplication of 11p15 associated with Silver-Russell syndrome? J Med Genet. 2005;42:e26. [PMC free article: PMC1736048] [PubMed: 15863658]
- Fisher AM, Thomas NS, Cockwell A, Stecko O, Kerr B, Temple IK, Clayton P. Duplications of chromosome 11p15 of maternal origin result in a phenotype that includes growth retardation. Hum Genet. 2002;111:290-6. [PubMed: 12215843]
- Fitzpatrick GV, Soloway PD, Higgins MJ. Regional loss of imprinting and growth deficiency in mice with a targeted deletion of KvDMR1. Nat Genet. 2002;32:426-31. [PubMed: 12410230]
- Flori E, Girodon E, Samama B, Becmeur F, Viville B, Girard-Lemaire F, Doray B, Schluth C, Marcellin L, Boehm N, Goossens M, Pingault V. Trisomy 7 mosaicism, maternal uniparental heterodisomy 7 and Hirschsprung's disease in a child with Silver-Russell syndrome. Eur J Hum Genet. 2005;13:1013-8. [PubMed: 15915162]
- Font-Montgomery E, Stone KM, Weaver DD, Vance GH, Das S, Thurston VC. Clinical outcome and follow-up of the first reported case of Russell-Silver syndrome with the unique combination of maternal uniparental heterodisomy 7 and mosaic trisomy 7. Birth Defects Res A Clin Mol Teratol. 2005;73:577-82. [PubMed: 16007591]
- Gicquel C, Rossignol S, Cabrol S, Houang M, Steunou V, Barbu V, Danton F, Thibaud N, Le Merrer M, Burglen L, Bertrand AM, Netchine I, Le Bouc Y. Epimutation of the telomeric imprinting center region on chromosome 11p15 in Silver-Russell syndrome. Nat Genet. 2005;37:1003-7. [PubMed: 16086014]
- Graham JM, Hoehn H, Lin MS, Smith DW. Diploid-triploid mixoploidy: clinical and cytogenetic aspects. Pediatrics. 1981;68:23-8. [PubMed: 6264378]
- Hannula K, Lipsanen-Nyman M, Kontiokari T, Kere J. A narrow segment of maternal uniparental disomy of chromosome 7q31-qter in Silver-Russell syndrome delimits a candidate gene region. Am J Hum Genet. 2001;68:247-53. [PMC free article: PMC1234921] [PubMed: 11112662]
- Joyce CA, Sharp A, Walker JM, Bullman H, Temple IK. Duplication of 7p12.1-p13, including GRB10 and IGFBP1, in a mother and daughter with features of Silver-Russell syndrome. Hum Genet. 1999;105:273-80. [PubMed: 10987657]
- Kim Y, Kim SS, Kim G, Park S, Park IS, Yoo HW. Detection of maternal uniparental disomy at the two imprinted genes on chromosome 7, GRB10 and PEG1/MEST, in a Silver-Russell syndrome patient using methylation-specific PCR assays. Clin Genet. 2005;67:267-9. [PubMed: 15691366]
- Kulkarni ML, Venkataramana V, Sureshkumar C, Shabeer HM. Russell-Silver syndrome: a study of 3 cases. Ann Dent. 1995;54:56-60. [PubMed: 8572550]
- Lai KY, Skuse D, Stanhope R, Hindmarsh P. Cognitive abilities associated with the Silver-Russell syndrome. Arch Dis Child. 1994;71:490-6. [PMC free article: PMC1030083] [PubMed: 7726606]
- Leppig KA, Saal HM, Simpson E, Disteche CM. Distal deletion of Yq in a patient with phenotype of Russell-Silver syndrome. Am J Hum Genet. 1991;49:301.
- Li CC, Chodirker BN, Dawson AJ, Chudley AE. Severe hemihypotrophy in a female infant with mosaic Turner syndrome: a variant of Russell-Silver syndrome? Clin Dysmorphol. 2004;13:95-8. [PubMed: 15057125]
- Li T, Hu JF, Qiu X, Ling J, Chen H, Wang S, Hou A, Vu TH, Hoffman AR. CTCF regulates allelic expression of Igf2 by orchestrating a promoter-polycomb repressive repressive complex 2 intrachromosomal loop. Mol Cell Biol. 2008;28:6473-82. [PMC free article: PMC2577414] [PubMed: 18662993]
- Martin RA, Grange DK, Zehnbauer B, Debaun MR. LIT1 and H19 methylation defects in isolated hemihyperplasia. Am J Med Genet A. 2005;134A:129-31. [PubMed: 15651076]
- Midro AT, Debek K, Sawicka A, Marcinkiewicz D, Rogowska M. Second observation of Silver-Russell syndrome in a carrier of a reciprocal translocation with one breakpoint at site 17q25. Clin Genet. 1993;44:53-5. [PubMed: 8403458]
- Monk D, Wakeling EL, Proud V, Hitchens M, Abu-Amero SN, Stanter P, Preece MA, Moore GE. Duplication of 7p11.2-p13, including GRB10. In Silver-Russell syndrome. Am J Hum Genet. 2000;66:36-46. [PMC free article: PMC1288348] [PubMed: 10631135]
- Moore GE, Abu-Amero S, Wakeling E, Hitchins M, Monk D, Stanier P, Preece M. The search for the gene for Silver-Russell syndrome. Acta Paediatr Suppl. 1999;88:42-8. [PubMed: 10626544]
- Netchine I, Rossignol S, Dufourg MN, Azzi S, Rousseau A, Perin L, Houang M, Steunou V, Esteva B, Thibaud N, Raux Demay MC, Danton F, Petriczko E, Bertrand AM, Heinrichs C, Carel JC, Loeuille GA, Pinto G, Jacquermont ML, Gicquel C, Cabrol S, Le Bouc Y. 11p15 ICR1 imprinting center region 1 loss of methylation is a common and specific cause of typical Russel-Silver syndrome: clinical scoring system and epigenetic-phenotypic correlations. J Clin endocrinol Metab. 2007;92:3148-54. [PubMed: 17504900]
- Noeker M, Wollmann HA. Cognitive development in Silver-Russell syndrome: a sibling-controlled study. Dev Med Child Neurol. 2004;46:340-6. [PubMed: 15132265]
- Obermann C, Meyer E, Prager S, Tomiuk J, Wollmann HA, Eggermann T. Searching for genomic variants in IGF2 and CDKN1C in Silver-Russell syndrome patients. Mol Genet Metab. 2004;82:246-50. [PubMed: 15234339]
- Orbak Z, Orbak R, Kara C, Kavrut F. Differences in dental and bone maturation in regions with or without hemihypertrophy in two patients with Russell-Silver syndrome. J Pediatr Endocrinol Metab. 2005;18:701-10. [PubMed: 16128247]
- Ortiz C, Cleveland RH, Jaramillo D, Blickman JG, Crawford J. Urethral valves in Russell-Silver syndrome. J Pediatr. 1991;119:776-8. [PubMed: 1658284]
- Ounap K, Reimand T, Magi ML, Bartsch O. Two sisters with Silver-Russell phenotype. Am J Med Genet A. 2004;131:301-6. [PubMed: 15523618]
- Partington MW. X-linked short stature with skin pigmentation: evidence for heterogeneity of the Russell-Silver syndrome. Clin Genet. 1986;29:151-6. [PubMed: 3955866]
- Pedreira CC, Savarirayan R, Zacharin MR. IMAGe syndrome: a complex disorder affecting growth, adrenal and gonadal function, and skeletal development. J Pediatr. 2004;144:274-7. [PubMed: 14760276]
- Price SM, Stanhope R, Garrett C, Preece MA, Trembath RC. The spectrum of Silver-Russell syndrome: a clinical and molecular genetic study and new diagnostic criteria. J Med Genet. 1999;36:837-42. [PMC free article: PMC1734267] [PubMed: 10544228]
- Ramírez-Dueñas ML, Medina C, Ocampo-Campos R, Rivera H. Severe Silver-Russell syndrome and translocation (17;20) (q25;q13). Clin Genet. 1992;41:51-3. [PubMed: 1633648]
- Reboul MP, Tadonnet O, Biteau N, Belet-de Putter C, Rebouissoux L, Moradkhani K, Vu PY, Saura R, Arveiler B, Lacombe D, Taine L, Iron A. Mosaic maternal uniparental isodisomy for chromosome 7q21-qter. Clin Genet. 2006;70:207-13. [PubMed: 16922723]
- Rizzo V, Traggiai C, Stanhope R. Growth hormone treatment does not alter lower limb asymmetry in children with Russell-Silver syndrome. Horm Res. 2001;56:114-6. [PubMed: 11847473]
- Russell A. A syndrome of intra-uterine dwarfism recognizable at birth with cranio-facial dysostosis, disproportionately short arms, and other anomalies (5 examples). Proc R Soc Med. 1954;47:1040-4. [PubMed: 13237189]
- Saal HM, Pagon RA, Pepin MG. Reevaluation of Russell-Silver syndrome. J Pediatr. 1985;107:733-7. [PubMed: 2414426]
- Saenger P. US experience in evaluation and diagnosis of GH therapy of intrauterine growth retardation/small-for-gestational-age children. Horm Res. 2002;58 Suppl 3:27-9. [PubMed: 12435893]
- Schönherr N, Meyer E, Roos A, Schmidt A, Wollmann HA, Eggermann T. The centromeric 11p15 imprinting centre is also involved in Silver Russell syndrome. J Med Genet. 2007;44:59-63. [PMC free article: PMC2597902] [PubMed: 16963484]
- Scott RH, Douglas J, Baskcomb L, Huxter N, Barker K, Hanks S, Craft A, Gerrard M, Kohler JA, Levitt GA, Picton S, Pizer B, Ronghe MD, Williams D., Factors Associated with Childhood Tumours (FACT) Collaboration. Cook JA, Pujol P, Maher ER, Birch JM, Stiller CA, Pritchard-Jones K, Rahman N2008Constitutional 11p15 abnormalities, including heritable imprinting center mutations, cause nonsyndromic Wilms tumor. Nat Genet 401329-34. [PubMed: 18836444]
- Shuman C, Steele L, Fei YL, Ray PN, Zackai E, Parisi M, Squire J, Weksberg R. Paternal uniparental disomy of 11p15 is associated with isolated hemihyperplasia and expands Beckwith-Wiedemann syndrome spectrum. Am J Hum Genet. 2002;71 Suppl:477.
- Silver HK, Kiyasu W, George J, Deamer WC. Syndrome of congenital hemihypertrophy, shortness of stature, and elevated urinary gonadotropins. Pediatrics. 1953;12:368-76. [PubMed: 13099907]
- Spengler S, Schönherr N, Binder G, Wollmann HA, Fircke-Otto S, Mühlenbert R, Denecke B, Baudis M, Eggermann T. Submicroscopic chromosome al imbalances in idiopathic Silver-Russell syndrome (SRS): the SRS phenotype overlaps with the 12q14 microdeletion syndrome. J Med Genet. 2010;47:356-60. [PubMed: 19762329]
- Stanhope R, Albanese A, Azcona C. Growth hormone treatment of Russell-Silver syndrome. Horm Res. 1998;49:37-40. [PubMed: 9716826]
- Tomiyama H, Ibuki T, Nakajima Y, Tanaka Y. Late intraoperative hypoglycemia in a patient with Russell-Silver syndrome. J Clin Anesth. 1999;11:80-2. [PubMed: 10396727]
- Toumba M, Albanese A, Azcona C, Stanhope R. Effect of long-term growth hormone treatment on final height of children with Russell-Silver syndrome. Horm Res Paediatr. 2010;74:212-7. [PubMed: 20424422]
- Turner CLS, Mackay DM, Callaway JLA, Docherty LE, Poole RL, Bullman H, Lever M, Castle BM, Kivuva EC, Turnpenny PD, Mehta SG, Mansour S, Wakeling EL, Verghese M, Madden J, Davies JH, Temple IK. Methylation analysis of 79 patients with growth restriction reveals novel patterns of methylation at imprinted loci. Eur J Hum Genet. 2010;18:648-55. [PMC free article: PMC2987339] [PubMed: 20104244]
- Vilain E, Le Merrer M, Lecointre C, Desangles F, Kay MA, Maroteaux P, McCabe ER. IMAGe, a new clinical association of intrauterine growth retardation, metaphyseal dysplasia, adrenal hypoplasia congenita, and genital anomalies. J Clin Endocrinol Metab. 1999;84:4335-40. [PubMed: 10599684]
- Wakeling EL, Amero A, Alders M, Bliek J, Forsythe E, Kumar S, Lim DH, MacDonald F, Mackay DJ, Maher ER, Moore GE, Poole RL, Price SM, Tangeraas T, Turner CLS, Van Haelst MM, Willoughby C, Temple IK, Cobben JM. Epigenotype-phenotype correlatinos in Silver-Russell syndrome. J Med Genet. 2010;47:760-8. [PMC free article: PMC2976034] [PubMed: 20685669]
- Weksberg R, Shuman C, Beckwith JB. Beckwith-Wiedemann syndrome. Eur J Hum Genet. 2010;18:8-14. [PMC free article: PMC2987155] [PubMed: 19550435]
- Wollmann HA, Kirchner T, Enders H, Preece MA, Ranke MB. Growth and symptoms in Silver-Russell syndrome: review on the basis of 386 patients. Eur J Pediatr. 1995;154:958-68. [PubMed: 8801103]
- Zeschnigk M, Albrecht B, Buiting K, Kanber D, Eggermann T, Binder G, Gromoll J, Prott EC, Seland S, Horsthemke B. IGF2/H19 hypomethylation in Silver-Russell syndrome and isolated hemihypoplasia. Eur J Hum Genet. 2008;16:328-34. [PubMed: 18159214]
建议阅读的文献资料
- Bruce S, Hannula-Jouppi K, Pouskari M, Fransson I. SImola KO, Lipsanen-Nyman M, Kere J. Submicroscopic genomic alterations in Silver-Russell syndrome and Silver-Russell-like patients. J Med Genet. 2010;47:816-22. [PubMed: 19752157]
- Hall JG. Review and hypothesis: syndromes with severe intrauterine growth restriction and very short stature—are they related to the epigenetic mechanism(s) of fetal survival involved in the developmental origins of adult health and disease? Am J Med Genet Part A. 2010;152A:512-27. [PubMed: 20101705]
- Rossignol S, Netchine I, Le Bouc Y. Epigenetics in Silver-Russell syndrome. Best Pract Res Clin Endocrinol Metab. 2008;22:403-14. [PubMed: 18538282]
- Schönherr N, Jäger S, Ranke MB, Wolmann HA, Binder G, Eggermann T. No evidence for isolated imprinting mutations in the PEG1/MEST locus in Silver-Russell patients. Eur J Med Genet. 2008;51:322-4. [PubMed: 18585117]
篇注
修订记录
- 2 June 2011 (me) Comprehensive update posted live
- 9 March 2007 (hs) Revision: 甲基化分析 for H19 clinically available
- 7 September 2006 (me) Comprehensive update posted to live Web site
- 5 March 2004 (me) Comprehensive update posted to live Web site
- 2 November 2001 (me) Review posted to live Web site
- February 2001 (hs) Original submission