
摘要
临床特征.
Citrin缺乏症在新生儿或婴儿中表现为由Citrin缺乏引起的新生儿肝内胆汁淤积(NICCD),在较大的儿童中表现为由Citrin缺乏引起的生长发育不良和血脂异常(FTTDCD),而在成人中表现为复发性高氨血症,并伴有瓜氨酸血症II型的神经精神症状 (CTLN2)。通常情况下,Citrin缺乏症的特点是强烈偏好富含蛋白质和/或脂质的食物,并排斥富含碳水化合物的食物。
- NICCD. 1岁以下的儿童有低出生体重史,伴有生长受限和暂时性肝内胆汁淤积、肝脏肿大、弥漫型脂肪肝、实质细胞浸润伴肝纤维化、可变肝功能不全、低蛋白血症、凝血因子减少、溶血性贫血,和/或低血糖。NICCD一般不严重,症状通常在一岁时通过适当的治疗可治愈,尽管在极少数情况下需要进行肝移植。
- FTTDCD. 超过一岁,许多患有Citrin缺乏症的患儿对富含蛋白质和/或脂肪的食物产生了偏好并排斥富含碳水化合物的食物。临床异常可能包括生长受限、低血糖、胰腺炎、重度疲劳、厌食和生活质量下降。实验室变化包括血脂异常、乳酸与丙酮酸比值增加、尿氧化应激标志物水平升高以及三羧酸(TCA)循环代谢物的严重偏差。数十年后,一些患有NICCD或FTTDCD的个体发展成CTLN2。
- CTLN2. 通常在20到50岁之间突然发病。表现为反复发作的高氨血症,伴有神经精神症状,包括夜间谵妄、攻击性、易怒、多动、妄想、定向障碍、不安、嗜睡、记忆丧失、震颤、惊厥发作和昏迷。这些症状通常是由酒精和糖的摄入、药物治疗和/或手术引起的。患者可能有或没有NICCD/FTTDCD病史。
诊断/检测.
Citrin缺乏症的诊断是建立在具有生化特征表现的个体(一般而言,血液或血浆中氨浓度升高,血浆或血清中瓜氨酸和精氨酸浓度升高,血浆或血清中苏氨酸与丝氨酸比值升高,血清中胰分泌胰蛋白酶抑制浓度升高)和鉴定SLC25A13双等位基因的 致病变异。
管理.
对症治疗: NICCD: 补充脂溶性维生素饮食和食用无乳糖和中链甘油三酯(MCT)丰富的配方。FTTDCD: 除饮食治疗外,服用丙酮酸钠可以改善生长发育。 CTLN2: 肝移植可预防高氨血症危象,纠正代谢紊乱,解除对富含蛋白质食物的偏好;精氨酸可降低血氨浓度,减少热量/碳水化合物摄入;增加蛋白质摄入可减少高甘油三酯血症。使用精氨酸、丙酮酸钠和MCT油可以延缓肝移植需求。
预防主要临床表现: 富含脂肪和蛋白质的低碳水化合物饮食。
监测: 定期测量与citrin缺乏症相关的血浆氨浓度和瓜氨酸浓度以及血清PSTI浓度。对患有NICCD的儿童进行随访,以获得FTTDCD的实验室和物理结果。
避免接触药物/环境: 低蛋白高碳水化合物饮食;甘油和果糖输注治疗脑水肿;酒精;对乙酰氨基酚和雷贝拉唑。
遗传咨询.
Citrin缺乏症的遗传方式为 常染色体隐性遗传。 若父母双方均为SLC25A13 致病性变异携带者,每一个子代同胞有25%概率为受累的个体,50%概率成为无症状携带者,25%概率非携带者也不受累。 当父母中的一方是携带者,另一方携带两个SLC25A13致病性变异时,受累个体的每个子代同胞有50%的概率受累,50%的概率成为无症状携带者。若家系成员携带有SLC25A13致病变异,可对高危亲属进行检测及高危妊娠进行产前诊断。
GeneReview 范畴
| Citrin缺乏症: 覆盖表型 1 |
|---|
|
同义词和既往名称参见 Nomenclature.
- 1.
其他相关表型的遗传因素参见 Differential Diagnosis.
诊断
Citrin缺乏症有两种公认的显著表型: citrin缺乏引起的新生儿肝内胆汁淤积 (NICCD) 和 瓜氨酸血症 II型 (CTLN2) (参见Figure 1) [Saheki & Kobayashi 2002, Yamaguchi et al 2002, Kobayashi & Saheki 2004, Saheki & Kobayashi 2005, Kobayashi et al 2006] – 和介于二者之间的第三个 表型: citrin缺乏引起的不能生长发育和血脂异常(FTTDCD) [Song et al 2011, Song et al 2013].
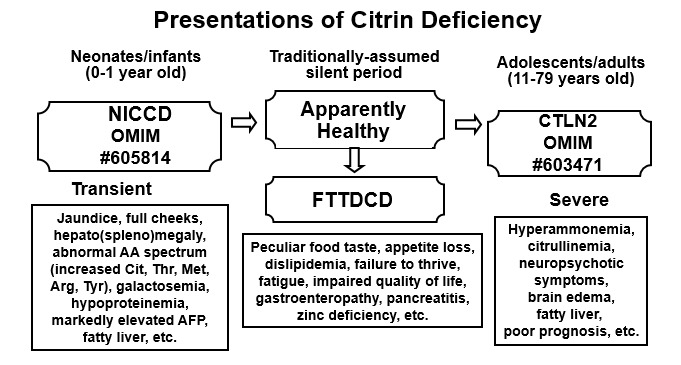
- citrin缺乏引起的新生儿肝内胆汁淤积 (NICCD)以短暂性新生儿胆汁淤积症和可变肝功能不全为特征。某些 受累的的个体因肝硬化预后不良。
- citrin缺乏引起的不能生长发育和血脂异常 (FTTDCD) 其特征是在CTLN2发作之前NICCD后生长受限,以及血脂浓度异常,包括甘油三酸酯,总胆固醇和HDL-胆固醇。在没有独特的食物偏好史或分子检测的情况下,很难在此阶段对Citrin缺乏症进行临床诊断。
- 瓜氨酸血症 II型 (CTLN2) 以儿童至成人反复发作的高氨血症和相关的神经精神症状为特征。
提示性发现
有以下表现的个体应怀疑为Citrin缺乏症 (参见 Figure 2):
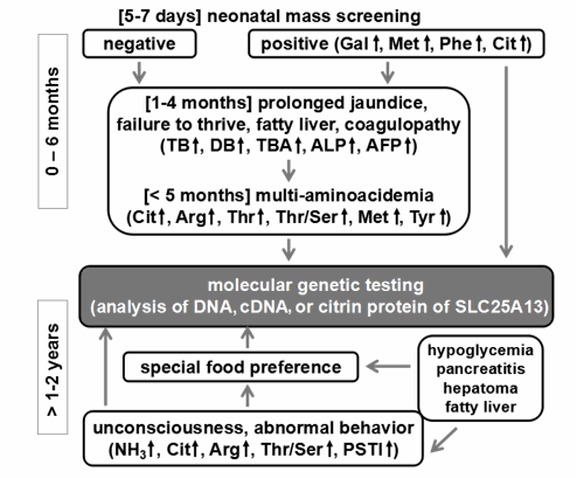
- 婴儿有一个积极的 新生儿筛查 测试:
- 瓜氨酸血症和/或长期黄疸;或
- 半乳糖血症,高甲硫氨酸血症,酪氨酸血症或高苯丙氨酸血症,在随访诊断检查中没有发现这些疾病。注意: NICCD患儿中约40%的新生儿血斑筛查中半乳糖,蛋氨酸和/或苯丙氨酸的血浆浓度升高 [Ohura et al 2003, Ohura et al 2007]. 尽管血浆酪氨酸在某些个体中升高,但这不是一个普遍特征。 相反,怀疑为酪氨酸血症,是由于在尿液GC-MS分析中4-羟苯基乳酸和4-羟苯基丙酮酸会增加。
- 一岁以上儿童生长发育不良和血脂异常
- 患有高氨血症的肝性脑病的年龄较大的儿童和成人,特别是有厌恶碳水化合物和喜爱富含蛋白质和脂肪食物的病史
- 不明原因复发性胰腺炎、高脂血症、脂肪肝或肝癌的儿童和成人
建立诊断
citrin缺乏症的诊断建立在生化检测结果和/或 分子遗传学检测鉴定SLC25A13 双等位基因的致病变异的先证者上(见Table 3)。 如果不能鉴定出致病性变异,则可以进行蛋白质印迹分析。
生化检测
下列检测结果进一步支持citrin缺乏症的诊断Table 1:
- 氨
- 氨基酸定量分析
- 胰腺分泌型胰蛋白酶抑制剂(PSTI)
Table 1.
Citrin缺乏症的生化表现
| 表型 (年龄) | 血液或血浆氨浓度 (µmol/L) | 血浆或血清浓度: | 血浆或血清苏氨酸与丝氨酸比值 | PSTI血清浓度 2 (ng/mL) | |
|---|---|---|---|---|---|
| 瓜氨酸 1 (µmol/L) | 精氨酸 (µmol/L) | ||||
| 对照 | 18-47 3 | 17-43 3 | 54-130 3 | 1.10 | 4.6-20 3 |
| NICCD (0-6 mos) | 60 | 300 | 205 | 2.29 | 30 |
| FTTDCD (>1-11 yrs) | 正常或轻度升高 | 正常或轻度升高 | 通常正常 | 未知 | 未知 |
| CTLN2 (11-79 yrs) | 152 | 418 | 198 | 2.32 | 71 |
PSTI = 胰腺分泌型胰蛋白酶抑制剂
- 1.
在新生儿筛查中发现瓜氨酸血症是NICCD最早发现的生化异常 [Tamamori et al 2004].
- 2.
由于部分NICCD患者的血清PSTI浓度较高 [Tamamori et al 2002],和 CTLN2发病前的患者血清PSTI浓度也较高[Tsuboi et al 2001],因此血清PSTI浓度的测定有助于CTLN2的症状前诊断。
- 3.
范围
Table 2.
NICCD患者0-6月龄血浆苏氨酸、蛋氨酸和酪氨酸的浓度
| 氨基酸 | 中值 (25%-75% Range) (µmol/L) | 参考值 (µmol/L) |
|---|---|---|
| 苏氨酸 | 496 (291-741) | 67-190 |
| 蛋氨酸 | 124 (53-337) | 19-40 |
| 酪氨酸 | 178 (99-275) | 40-90 |
分子遗传学检测
分子遗传学检测方法包括单基因检测和使用多基因面板检测。
单基因测试. 如果仅有1个或没有发现致病性变异 ,首先考虑SLC25A13序列分析和其次考虑基因-靶向缺失/重复分析。
致病性变异的靶向分析可以首先在日本或中国血统的个体中进行。
- 在日本citrin缺乏症患者中,2种致病性变异占致病等位基因的绝大多数(约70%)(参见Molecular Genetics,致病性变异)。
- [Lin et al 2016] (see Molecular Genetics, Pathogenic variants).在中国citrin缺乏症患者中,四种致病性变异占致病等位基因的80%以上[Lin et al 2016] (参见Molecular Genetics,致病性变异)。
A multigene panel 多基因面板考虑包括SLC25A13和其他相关基因 (参见 Differential Diagnosis). 注意: (1) 面板中包含的基因和每个基因检测的诊断敏感性 因实验室而异,并可能随时间而变化(2) 一些多基因面板可能包含与本GeneReview中讨论的病症无关的基因; 因此,临床医生需要以最合理的成本确定哪个多基因面板最有可能确定该病的遗传病因,同时限制识别对无法解释潜在表型的意义不确定的基因中的致病变异。(3) 在某些实验室中,基因面板选线可能包括实验室设计定制的面板和/或定制以表型为重点的外显子组分析,其中包括临床医生指定的基因。 (4) 面板中使用的方法可能包括序列分析,缺失/重复分析和/或其他基于非序列的检测。
由于SLC25A13中与该疾病相关的大片段缺失或重复无法通过序列分析检测到,因此建议多基因面板也应包括缺失/重复分析(参见 Table 3)。
关于多基因面板的更多介绍请点击 here. 有关制定基因检测的临床医生的更多详细信息,请参见 here.
Table 3.
缺乏症分子遗传学检测
| 基因 1 | 通过该方法检出致病性变异2的先证者个体 | |
|---|---|---|
| SLC25A13 | 序列分析 3 | 85%-90% 4 |
| 基因-靶向缺失/重复 5 | 10%-15% 6 |
- 1.
- 2.
该基因等位基因变异信息参见Molecular Genetics
- 3.
- 4.
- 5.
- 6.
Western Blot Analysis免疫印迹分析
对于citrin蛋白的免疫印迹分析,可以考虑在生化检测中没有发现或 分子遗传学检测 只找到一个SLC25A13 致病性变异的罕见情况下使用。
使用氨基端特异性抗人citrin抗体的免疫印迹分析在肝脏或培养的 双等位基因的SLC25A13致病性变异个体的成纤维细胞中很少检测到或没有交叉反应性免疫物质[Takahashi et al 2006, Dimmock et al 2007, Fu et al 2011]. 外周血淋巴细胞可以作为免疫印迹分析的另一个样本来源 [Tokuhara et al 2007], 从培养的淋巴细胞中提取线粒体蛋白,免疫印迹分析能更容易地检测到citrin蛋白[Zhang et al 2017].
临床特征
临床描述
Citrin缺乏症可表现为新生儿或婴儿由Citrin缺乏引起的新生儿肝内胆汁淤积症(NICCD), 在年龄较大的儿童中,由于缺乏citrin可导致生长发育不良和血脂异常 (FTTDCD), 在成年人中表现为复发性高氨血症伴神经精神症状瓜氨酸血症II型的(CTLN2). FTTDCD和CTLN2通常以个人偏爱富含蛋白质和/或富含脂质的食物以及对富含碳水化合物的食物为特征。患有CTLN2的个体可能有或没有NICCD或FTTDCD病史。NICCD或FTTDCD患者发展为CTLN2的比例尚不清楚。 对于那些除了饮食管理之外没有特殊医学问题的NICCD患者,建议进行密切的医学观察,因为他们即使在疾病静止期 [Nagasaka et al 2009, Nagasaka et al 2017]仍有生化改变 [Song et al 2011]。
Citrin缺乏引起的新生儿肝内胆汁淤积症 (NICCD)
年龄小于1岁的NICCD患儿有短暂性肝内胆汁淤积(参见 Table 4). 其他表现包括弥漫性脂肪肝伴肝肿大和肝纤维化相关的实质细胞浸润,低出生体重史,生长受限,低蛋白血症,凝血因子减少,溶血性贫血,可变(主要是轻度)肝功能障碍,和/或低血糖。
Table 4.
MNICCD 0-6月龄肝细胞功能测定
| 检验项目 | 中值 (25%-75% range) (mg/dL) | 参考范围 (mg/dL) |
|---|---|---|
| TB in NICCD | 4.9 (2.8-8.0) | 0.2-1.0 |
| DB in NICCD | 2.5 (1.5-3.7) | 0-0.4 |
| TB/DB ratio in NICCD | 0.55 (0.41-0.66) | — |
| TBA | 239 (172-293) | 5-25 |
| AFP | 91,900 (33,200-174,700) | 260-6,400 1, 2 2-55 2, 3 |
TB = 总胆红素
DB = 直接胆红素
TBA = 血清总胆汁酸
AFP = 甲胎蛋白
- 1.
0-1 月
- 2.
- 3.
>1 月
NICCD一般不严重,尽管在少数情况下需要肝移植 [Tamamori et al 2002, Kobayashi et al 2006]. 经过一年的治疗,包括补充脂溶性维生素和使用无乳糖治疗配方(用于继发性半乳糖血症患者)和/或富含中链甘油三酯(MCT)的治疗配方,症状通常会随着年龄的增长而缓解 [Ohura et al 2003, Song et al 2010, Hayasaka et al 2012, Zhang et al 2014a].
从一到两岁左右开始,孩子们就对富含蛋白质和脂肪的食物表现出强烈嗜好,而对富含糖分和碳水化合物的食物则反感排斥 [Hachisu et al 2005, Saheki & Kobayashi 2005, Saheki et al 2008].
在第二个或随后的几十年中,一些citrin缺乏症患者会发展为严重的CTLN2并伴有神经精神症状 [Saheki & Kobayashi 2002]. 通常,从NICCD之后的适应(和/或补偿)阶段过渡到CTLN2的发作是进行性的,但是CTLN2的表现通常是突发的。
Citrin缺乏引起的生长发育不良和血脂异常 (FTTDCD)
FTTDCD近来被提出作为CTLN2发病前的新表型 [Song et al 2011]. FTTDCD的临床和实验室特征仍在构建中。在这一时期(传统上认为是CTLN2发病前的“表面健康”阶段),一些患儿发现有实验室和/或临床异常。
实验室异常包括血脂异常,表现为甘油三酸酯和总胆固醇和低密度脂蛋白胆固醇水平较高,而高密度脂蛋白胆固醇水平较低[Song et al 2009a, Song et al 2011] 和其他发现,如乳酸-丙酮酸比值增加,尿氧化应激标志物水平升高,三羧酸(TCA)循环代谢物显著偏差 [Kobayashi & Saheki 2004, Saheki & Kobayashi 2005, Kobayashi et al 2006, Nagasaka et al 2009, Lee et al 2010, Takeuchi et al 2015, Nagasaka et al 2017].
临床异常包括生长受限、低血糖和胰腺炎。在适应和补偿阶段(传统上假定为“沉默”)citrin缺乏的患儿中发现严重疲劳和生活质量受损 [Okano et al 2013]. 此外,据报道,一名12岁患有citrin缺乏症的女性,表现为严重的厌食症和体重减轻,类似于限制型神经性厌食症 [Takeuchi et al 2015].
瓜氨酸血症 II 型(CTLN2)
CTLN2的特点是反复出现高氨血症和神经和精神症状,与肝性脑病或遗传性尿素循环代谢紊乱非常相似,包括夜间谵妄、异常行为(攻击性、易怒和多动性)、妄想、定向障碍、烦躁不安、嗜睡,失忆、震颤、抽搐和昏迷。脑CT正常,脑电图呈弥漫性慢波。
表现为突发性,通常发生在20岁至50岁之间(范围:11-79岁;平均年龄:34.4±12.8岁;n = 103) [Yasuda et al 2000].
许多患有CTLN2的人对富含蛋白质和/或富含脂质的食物(如豆类、花生、鸡蛋、牛奶、奶酪、鱼和肉)有强烈嗜好,强烈排斥富含碳水化合物的食物(包括大米、果汁和糖果)。症状通常由酒精和糖的摄入、药物治疗和/或手术引起。
大多数人都很瘦。超过90%的人体重指数低于20,约40%的人体重指数低于17(范围:15.6-19.1;n = 110) [Kobayashi et al 2006] (日本健康个体范围:男性20-24;女性19-23).
以下并发症发生在10%以上的CTLN2患者中 [Kobayashi et al 2000].
- 胰腺炎. 青少年型慢性胰腺炎和无肝硬化的肝细胞癌可先于CTLN2出现 [Ikeda et al 2004].
- 高脂血症. 如果为citrin缺乏症患者提供高碳水化合物的餐食,通常会观察到高甘油三酸酯血症[Imamura et al 2003].
- 脂肪肝. 大多数患有NICCD和CTLN2的人都患有脂肪肝,其组织学特征与NASH(非酒精性脂肪性肝炎)相同。 [Takagi et al 2006, Fukumoto et al 2008, Komatsu et al 2008]. 轻度纤维化也可见 [Kobayashi et al 2000].
肝内胆汁淤积症是罕见的; 然而,回顾分析发现有些人在儿童早期就患有NICCD征象 [Kobayashi & Saheki 2004, Saheki & Kobayashi 2005]. 例如,一名接受肝移植的16岁CTLN2患者[Kasahara et al 2001] 在婴儿早期曾出现过短暂性低蛋白血症和黄疸 [Tomomasa et al 2001]。
实验室检查异常结果
- 血浆或血清中的Fischer比(支链氨基酸[BCAAs]Val+Leu+Ile/芳香族氨基酸[AAAs]Tyr+Phe)从~3.4下降到~2。
- 肝脏特异性精氨酸琥珀酸合成酶(ASS)酶活性降低至对照组的10%左右(SLC25A13中致病变异的次要影响) [Yasuda et al 2000].
- 几乎所有CTLN2患者的血浆中,血浆中甲胎蛋白的浓度都是正常的 [Kobayashi et al 1997] ,除了一些与肝癌相关的CTLN2患者 [Hagiwara et al 2003]。
病理结果 包括肝脏脂肪浸润和轻度纤维化,几乎没有肝功能障碍。
基因型-表型相关性
目前尚不明确该疾病的基因型-表现型相关性。
外显率
- NICCD的男女比例大致相等(73:80),而CTLN2的男女比例为2.4:1 (120:50)[Kobayashi & Saheki 2004].
命名
NICCD. NICCD病因在确认为 双等位基因的SLC25A13致病变异之前被称为“不明原因的原发性新生儿肝炎伴脂肪肝”[Ohura et al 1997]在证实存在SLC25A13致病性变体之前。
CTLN2.Miyakoshi et al [1968] 据报道血浆瓜氨酸浓度在高氨血症和独特的慢性复发性肝脑退行性变患者中升高。这种肝脑变性以脑病理改变为基础,被称为“假性胆管性肝脑疾病”;以饮食高度不均衡引起的代谢障碍或内分泌异常引起的发育障碍为基础,被称为“营养性肝脑疾病”。
Saheki et al [1981]据报道,这种肝性脑病是瓜氨酸血症的一种,伴精氨酸琥珀酸合成酶活性/蛋白定性和肝脏特异性降低,后来 Saheki et al [1985] 将其命名为“成人发作的瓜氨酸血症II型”
流行病学
在日本,SLC25A13致病变异的纯合子或复合杂合子的患病率根据 携带者 (杂合子)1:65 [Saheki & Kobayashi 2002, Tabata et al 2008]的比率计算为1:17000。这与观察到的NICCD患病率相似,但与CTLN2的患病率(1:100000-1:230000)不同[Kobayashi et al 2006]。根据他们的观察,作者认为大多数具有 双等位基因的SLC25A13致病个体的日本血统去群体都患有NICCD。
直到最近,人们还认为citrin缺乏症仅限于日本;citrin缺乏症现在被认为是广泛民族性的[Dimmock et al 2009]. 在以色列、巴基斯坦、美国、英国、中国和捷克共和国已经发现了具有新的SLC25A13致病变异个体 [Ben-Shalom et al 2002, Hutchin et al 2006, Luder et al 2006, Dimmock et al 2007, Fiermonte et al 2008, Song et al 2008, Tabata et al 2008, Song et al 2009b, Song et al 2011].
中国携带者频率为(1:65),特别是在男方地区,包括台湾 (1:48)和韩国朝鲜 (1:112)。 [Lu et al 2005, Lee et al 2011].
遗传相关(等位基因)疾病
CTLN2, NICCD, 和 FTTDCD 是目前已知与致病变异相关的表型。
鉴别诊断
瓜氨酸血浆浓度, citrin缺乏症血浆瓜氨酸浓度增加, 在以下疾病瓜氨酸浓度亦增加:
- 瓜氨酸血症1型(CTLN1;ASS缺陷). CTLN1表现为广泛的重叠表型:急性新生儿型(“经典型”)、轻度晚发型(“非经典型”)、无症状或高氨血症型,以及女性怀孕或产后重症发作型 [Gao et al 2003].
- 新生儿(“经典”)型. 出生后不久,急性新生儿型的婴儿会出现高氨血症及其并发症,在没有及时干预的情况下死亡。及时接受治疗的患者可能存活一段时间,但通常伴有严重的神经功能缺陷
- 非经典型. 在晚发型患者中,高氨血症的发作与急性新生儿型相似,但早期的神经学表现轻微.
CTLN1是由于ASS酶缺乏所致,ASS是尿素循环的第三步,即瓜氨酸与天冬氨酸缩合形成精氨酸琥珀酸。未经治疗的CTLN1患者出现高氨血症,血浆瓜氨酸浓度升高,血浆精氨酸浓度降低。CTLN1是由ASS1 双等位基因的 致病变异引起的,并以常染色体隐性遗传。(注:在CTLN2中,由于肝脏ASS mRNA或ASS1无异常,因此肝脏ASS蛋白的特异性缺陷源于未知机理 [Yasuda et al 2000] )通过生化实验室检测(包括发现低血浆精氨酸)可将这种情况与citrin缺乏症鉴别. - 精氨琥珀酸尿症 (精氨琥珀酸裂解酶[ASL]缺乏) (参见 Urea Cycle Disorders Overview)
- 肾功能不全
- Classic galactosemia典型的半乳糖血症. 在一名新生儿中,典型的半乳糖血症表现为citrin缺乏 [Feillet et al 2008].
高氨血症, 见于 citrin缺乏症,也发生在尿素循环障碍中,这是由蛋白质和其他含氮分子分解产生的氮代谢缺陷引起的 (see Urea Cycle Disorders Overview).尿素循环中前四种酶(CPSI、OTC、ASS、ASL)、鸟氨酸转运体或辅助因子生成(NAGS)中的任何一种严重缺乏或活性完全缺失,会导致大多数受累的个体在生命的最初几天内氨和其他前体代谢物积累.
新生儿/婴儿胆汁淤积症, 见于citrin缺乏症,也发生在以下疾病中:
- 特发性新生儿肝炎(INH)与肝外胆管闭锁(EBA).与INH和EBA相比,NICCD血清直接胆红素或ALT水平较低,血清总胆汁酸和碱性磷酸酶水平较高。与INH相比,NICCD患者血清γ-GTP水平升高,AST活性降低. [Tazawa et al 2005].
- 进行性家族性肝内胆汁淤积症 (PFIC). NICCD高血清γ-GTP水平可与其他γ-GTP水平正常低值的肝内胆汁淤积症(PFIC)和良性复发性肝内胆汁淤积症(BRIC)相鉴别。PFIC1是由ATP8B1中 双等位基因的致病变异引起的(参见ATP8B1 Deficiency); PFIC2是由ABCB11(OMIM 601847)中的双等位基因致病变异引起的。 某些BRIC病例是由ATP8B1中的双等位基因致病变异引起的。
遗传性黄疸和高胆红素血症 是由胆红素代谢缺陷引起的。这些疾病主要包括非结合(间接)高胆红素血症(UDP-葡萄糖醛酸转移酶1-1缺陷)和结合(直接)高胆红素血症(小管ATP依赖性转运体缺陷:ABCC2[OMIM 601107]、ABCB11[OMIM 603201]或ATP8B1[ATP8B1 Deficiency])
其他
- 门体分流可通过血管造影排除。这些分流是指血流从门静脉流向下腔静脉、肝静脉或脾静脉。citrin缺乏症患者在影像学检查 (e.g., sonography, MRI).中没有类似分流.
- 超过30%的CTLN2患者最初被误诊为癫痫发作和/或心理障碍(如抑郁症、精神分裂症);其他人可能被诊断为患有肝癌、胰腺炎和高脂血症等疾病.
管理
初步诊断后的评估
为确立citrin缺乏症个体诊断需求和疾病表型程度,建议评估以下方面:
NICCD
- 评估肝脏和脾脏的大小.
- 通过腹部超声、CT或MRI寻找脂肪肝证据.
- 调研喂养方式.
FTTDCD
- 使用年龄性别相匹配的生长标准进行详细的人体测量检查和评估.
- 调研喂养方式.
CTLN2. 调研饮食中的碳水化合物、蛋白质和脂类成分.
所有表型. 咨询临床遗传学家和/或遗传咨询师.
对症治疗
NICCD. 在补充脂溶性维生素和使用不含乳糖和富含MCT的治疗配方后,大多数NICCD患儿的症状在12个月大时就会缓解消失 [Ohura et al 2003, Song et al 2010, Hayasaka et al 2012, Zhang et al 2014a].
两个同胞在从母乳改为含脯氨酸含量较高的配方奶后有所改善 [Ben-Shalom et al 2002].
一些患有NICCD的儿童无需治疗即可改善症状,这可能是减少母乳和/或普通配方食品的效果,同时引入富含蛋白质和脂质的固体补充剂,例如鸡蛋和肉类,因此对citrin缺乏症的个体有益 [Song et al 2010].
用治疗配方进行治疗并不是终生的. 大多数患有NICCD的婴儿在一岁时在临床和生化方面恢复,此时可摄入富含蛋白质和脂质质地或固体补充剂。目前尚不清楚继续治疗一年以上是否可以减少FTTDCD和CTLN2 表型 的可能性。.
此外,缺锌在NICCD中很常见,因此,当实验室证据表明缺锌时,应鼓励补锌,特别是在发育不良的个体中。
4例NICCD和严重肝功能不全的婴儿被诊断为不明原因的酪氨酸血症,并在10至12个月大时接受了肝移植 [Tamamori et al 2002, Kobayashi et al 2006].
FTTDCD. 对于这种新型citrin缺乏症表型,治疗措施描述很少.
- 患有FTTDCD的幼儿根据他自己的食物偏好(包括厌恶米饭和嗜好鱼)进行喂食;FTT逐渐改善,三岁时体重恢复超过第三百分位.血脂异常也逐渐好转 [Song et al 2009a].
- 除饮食治疗外,服用丙酮酸钠可能有效纠正生长受限 [Mutoh et al 2008, Saheki et al 2010]. .丙酮酸钠降低肝细胞中NADH/NAD+比值,这是citrin缺乏症发展的关键改变,可能与其改善生长受限有关。
CTLN2. 迄今为止,最成功的治疗方法是肝移植 [Ikeda et al 2001, Kasahara et al 2001, Yazaki et al 2004, Hirai et al 2008],该疗法可预防突发性高氨血症危象,纠正代谢紊乱,并消除对富含蛋白质食物的偏好 [Kobayashi & Saheki 2004]。 过去几乎所有CTLN2患者都需要肝移植;然而,精氨酸、丙酮酸钠和中链甘油三酯(MCT)油的引入改变了这种情况。其他治疗方法包括以下方面:
- 据报道,精氨酸(5-10 g/天)可有效降低血氨浓度.
- 减少热量/碳水化合物的摄入和增加蛋白质的摄入可以改善高甘油三酯血症 [Imamura et al 2003].
- 丙酮酸钠(4-9g/天)可有效降低高氨血症发作的频率,并改善一些病例的生长发育 [Mutoh et al 2008; Saheki et al 2010; Yazaki et al 2010; Ohura et al, personal communication; Okano et al, personal communication].
- 服用MCT油(含85%MCT的Mactone油; 45 mL /天)与所有正常实验室检查结果完全恢复或无高氨血症症状改善相关 [Hayasaka et al 2014].
首发表现预防
为防止高氨血症和促进生长,建议食用富含蛋白质和脂质、低碳水化合物的食物 [Saheki & Kobayashi 2005, Saheki et al 2006, Dimmock et al 2007, Saheki et al 2008, Dimmock et al 2009].
应避免高碳水化合物食物和酒精.
精氨酸可有效预防高氨血症危象.
预防继发并发症
维生素D缺乏和锌缺乏是NICCD的常见并发症 [Song et al, personal communication]. 据报道严重感染和肝硬化为一些NICCD患者的致命性并发症。因此,建议NICCD患者补充维生素D和锌并积极控制感染。
监测
为了监测一岁以上citrin缺乏症患者是否出现FTTDCD表型,应密切监测人体测量指标(如身高、体重和头围;血脂水平包括甘油三酯、总胆固醇、高密度脂蛋白胆固醇和低密度脂蛋白胆固醇)。
建议每隔几个月进行以下监测:
- 血浆氨浓度(特别是在晚上或喂食后2小时)
- 血浆瓜氨酸浓度
- 血清PSTI浓度
血浆瓜氨酸浓度和血清PSTI的升高提示CTLN2的发作 [Tsuboi et al 2001, Mutoh et al 2008] 应及时开始治疗。
媒介/环境避免接触
低蛋白/高热量(高碳水化合物)饮食. 尽管低蛋白/高热量饮食有助于预防尿素循环酶缺乏症中的高氨血症,但对患有各种形式citrin酸缺乏症的个体均有害 (i.e., NICCD, FTTDCD, or CTLN2) [Saheki et al 2004, Saheki & Kobayashi 2005, Saheki et al 2006]. 高碳水化合物饮食可能会增加NADH的产生,干扰尿素的合成并刺激柠檬酸-苹果酸的穿梭,从而导致高氨血症,脂肪肝和高甘油三酸酯血症 [Saheki & Kobayashi 2002, Imamura et al 2003, Saheki et al 2006, Saheki et al 2007].
输注糖,包括甘油,果糖和葡萄糖. 用含甘油的渗透剂治疗严重脑水肿可导致病情持续恶化,CTLN2患者禁用 [Yazaki et al 2005].大量的甘油和果糖的降解在肝细胞质中产生NADH,可能会干扰肝功能 [Saheki et al 2004, Yazaki et al 2005, Takahashi et al 2006].
输注高浓度葡萄糖也可能加剧高氨血症 [Tamakawa et al 1994, Takahashi et al 2006].
注:甘露醇注射液似乎更安全 [Yazaki et al 2005].
酒精. 饮酒可触发CTLN2发作,因为酒精脱氢酶(ADH)在肝细胞质中生成NADH.
药物.对乙酰氨基酚和雷贝拉唑可引发CTLN2 [Shiohama et al 1993, Imamura et al 2003].
亲属患病风险评估
在症状出现之前对婴儿进行适当的饮食管理(停止母乳喂养和引入无乳糖和/或富含MCT的配方食品),在确认citrin缺乏症先证者的高危无症状同胞的遗传状况下是合适的。
由于citrin缺乏症的无症状或有症状的个体并不总是表现出生化异常,因此对有风险的亲属(例如同胞)的确切诊断将在很大程度上取决于先证者中的SLC25A13分子遗传学发现:
- 如果在先证者病例中已明确两个SLC25A13致病变异,分子遗传学检测可有效使用。
- 如果先证者只有确认一个SLC25A13 致病性变异的情况下,已确认一个致病变异的高危亲属无法明确排除诊断。在这种情况下,需要经过随访一段时间的一系列临床和生化评估来确认或排除诊断。
- 如果先证者没有确认SLC25A13 致病性变异情况下,对高危人群进行分子检测将无济于事。在这种情况下,需要经过随访一段时间的一系列临床和生化评估来确认或排除诊断
参见Genetic Counseling,以了解有关高危亲属检测的遗传咨询问题。
在研疗法
搜索美国的ClinicalTrials.gov和欧洲的 www.ClinicalTrialsRegister.eu,可以获得关于各种疾病和条件的临床研究信息。注意:对于该疾病可能没有临床试验。
遗传咨询
遗传咨询是一个给患者及家属提供关于遗传性疾病本质、遗传特性以及影响并帮助他们做出知情的医疗决定的过程。下列段落描述遗传风险的评估以及根据家族史和基因检测判断家族成员遗传状态。本段落描述不适用于解决患者实际面对的个人、文化或伦理问题,也不能代替专业的遗传咨询。. —ED.
遗传模式
Citrin缺乏症遗传模式为常染色体隐性遗传 。
家庭成员风险
先证者父母
- 父母可能会有两个SLC25A13致病变异,但没有CTLN2的严重症状 [Kobayashi et al 2006],在163个患有NICCD的日本家庭中,48个父亲中有2个以及54个母亲中有1个均发现这一现象。一位无症状的父亲与其患有NICCD的儿子有相同的SLC25A13基因型 [Zeng et al 2014].
- 杂合子(携带者)无症状。
先证者同胞
- 总的来说,遗传SLC25A13致病性变异的同胞会受累并表现出citrin缺乏症的临床表现,与家系中的 先证者表型相似;然而,观察到的家族内临床外显率降低和家族内临床异质性。[Kobayashi & Saheki 2004; Kobayashi et al 2006; Zeng et al 2014; Okano, personal communication]
- 杂合子(携带者)无症状.
先证者子代. 除非 citrin缺乏症患者有一个受累的或携带者子代,否则他/她的子代将是SLC25A13 致病性变异的专性杂合子(携带者)。
携带者(杂合子)检测
对风险亲属进行携带者检测,需要事先在家族中确认SLC25A13致病变异。
遗传咨询相关问题
有关评估高危亲属以进行早期诊断和治疗的信息,请参阅管理、Evaluation of Relatives at Risk。
家庭计划
- 确定遗传风险、携带者 情况以及探讨产前检测的最佳时机是怀孕前。
DNA 库用于存储DNA(通常为提取自白细胞DNA)已备将来之需,我们对基因、等位基因突变的理解、对疾病的认识以及检测方法都将提升,应考虑对患者 受累的个体DNA进行保存。
产前检测和遗传学植入前诊断
一旦在一个受累的家庭成员中确认SLC25A13致病性变异,对风险增加的妊娠进行产前检测和植入前遗传学诊断是可行的。
在应用产前检测方面,医学专业人员和家庭内部可能存在不同的看法,特别是在考虑产前检测是为了终止妊娠而不是进行早期诊断的情况下。尽管大多数中心会考虑由父母决定是否进行产前检测,但对这些问题的讨论是合理的。
资源
GeneReviews 工作人员已经筛选了以下专科疾病和患者帮扶组织,注册登记能使患者及其家庭获益。GeneReviews对该类组织提供的信息不负责,信息的筛选标准,参见此处here
- National Library of Medicine Genetics Home Reference
- Children's Liver Disease Foundation (CLDF)36 Great Charles StreetBirmingham B3 3JYUnited KingdomPhone: +44 (0) 121 212 3839Fax: +44 (0) 121 212 4300Email: info@childliverdisease.org
- Metabolic Support UK5 Hilliards Court, Sandpiper WayChester Business ParkChester CH4 9QPUnited KingdomPhone: 0845 241 2173Email: contact@metabolicsupportuk.org
分子遗传学
分子遗传学检测的信息和OMIM相关列表可能同其它GeneReview信息会有不同。(可能有更新原因). —ED.
Table A.
Citrin缺乏症: 基因和数据库
| 基因 | 染色体位置 | 蛋白 | 基因座特异性数据库 | HGMD | ClinVar |
|---|---|---|---|---|---|
| SLC25A13 | 7q21 | Calcium-binding mitochondrial carrier protein Aralar2 | SLC25A13 @ LOVD | SLC25A13 | SLC25A13 |
Table B.
OMIM收录的Citrin缺乏症词条 (View All in OMIM)
基因结构.SLC25A13由18个外显子组成 [Kobayashi et al 1999, Sinasac et al 1999]. 基因和蛋白质信息的详细摘要, 参见 Table A, 基因.
致病性变异. 迄今为止,在外显子或内含子中已有超过100种致病性变异,致病性错义突变、预测的 citrin截短突变或异常的mRNA剪接[Kobayashi et al 1999, Lin et al 2012, Takahashi et al 2012, Chen et al 2013, Song et al 2013, Tong et al 2013, Wen et al 2013, Zeng et al 2014, Zhang et al 2014a, Zhang et al 2014b, Bijarnia-Mahay et al 2015, Wang et al 2015, Xiong et al 2015, Lin et al 2016, Zeng et al 2016, Zheng et al 2016, Oh et al 2017, Zhang et al 2017].
- 在日本citrin缺乏症患者中,两个致病性变异(c.1177 + 1G> A和c.851-854del)占致病等位基因的大部分(〜70%)。
- 在来自264个中国家庭的274名citrin缺乏症患者中,四个致病性变异(c.851-854del,c.615 + 5G> A,c.1750 + 72_1751-4dup17ins NM_138459.3:2667和c.1638_1660dup23) 占致病性等位基因的84.47% [Lin et al 2016].
在日本人中鉴定出的20种致病性变异中,有一些是在中国,越南和韩国的citrin缺乏症(NICCD或CTLN2)中发现的。 [Lu et al 2005, Lee et al 2006, Song et al 2006, Tsai et al 2006, Yeh et al 2006, Ko et al 2007a, Ko et al 2007b, Song et al 2008, Tabata et al 2008].
Different pathogenic variants were found in Israel, the United States, the United Kingdom, and China [Ben-Shalom et al 2002, Hutchin et al 2006, Luder et al 2006, Dimmock et al 2007, Song et al 2008, Tabata et al 2008, Song et al 2009b, Xing et al 2010, Fu et al 2011, Song et al 2011].
Table 5.
SLC25A13 致病性变异选取列表
| DNA核苷酸改变 (Alias 1) | 预测蛋白改变 | 参考序列 | 参考来源 |
|---|---|---|---|
| c.615+5G>A (IVS6+5G>A) | -- | NM_014251 NP_055066 | Saheki et al [2004] |
| c.851_854del (851del4) | p.Met285ProfsTer2 | Kobayashi et al [1999] | |
| c.1078C>T | p.Arg360Ter | Tabata et al [2008] | |
| c.1177+1G>A (IVS11+1G>A) | -- | Kobayashi et al [1999] | |
| c.1638_1660dup23 (1638ins23) | p.Ala554GlyfsTer17 | Kobayashi et al [1999] | |
| c.1750+72_1751-4dup17ins NM_138459.3:2667 2 (IVS16ins3kb) | -- | Tabata et al [2008] |
关于变异分类的注意事项:表中列出的变异由作者提供。 GeneReviews工作人员尚未独立校验变异分类。
命名法注释:GeneReviews遵循人类基因组变异学会的标准命名 (varnomen
- .hgvs.org ). 参见 Quick Reference 关于命名解释.- 1.
变异名称不符合当前命名法规则。
- 2.
NUS1在6q22.31反义方向上 插入2667个互补核苷酸的复杂等位基因(参考序列 NM_138459
- .3 ); 在第16内含子的两侧插入17个核苷酸重复(NM_014251- .2 :c.1751-4_-22dup17) [Tabata et al 2008].
正常基因产物. Citrin及其同系物aralar[del Arco & Satrústegui 1998]是SLC25(溶质载体家族25)蛋白家族的成员。 这两种蛋白均位于线粒体内膜,作为苹果酸-天冬氨酸-NADH穿梭的组成部分,作为Ca2+结合/刺激的天冬氨酸谷氨酸载体(AGC)发挥作用 [Palmieri et al 2001, Kobayashi & Saheki 2003].
在肝脏中表达,aralar在脑和骨骼肌中表达,二者均在肾脏和心脏表达 [Kobayashi et al 1999]. Citrin作为肝脏AGC在各种代谢途径中起作用,包括有氧糖酵解,糖异生,尿素循环以及蛋白质和核苷酸合成 [Saheki & Kobayashi 2002, Saheki et al 2004, Saheki & Kobayashi 2005, Saheki et al 2006].
异常 基因产物. 大多数SLC25A13致病性变异会导致或预测citrin蛋白的截短或缺失线粒体跨膜结构域之间的一个环。使用一半抗人体citrin蛋白N端抗体的免疫印迹分析证实citrin蛋白严重缺乏,在具有双等位基因的SLC25A13致病性个体中,肝脏、培养的成纤维细胞和淋巴细胞中检测不到或几乎没有交叉反应的免疫物质 [Yasuda et al 2000, Takahashi et al 2006, Dimmock et al 2007, Tokuhara et al 2007, Fu et al 2011].
参考文献
引用文献
- Ben-Shalom E, Kobayashi K, Shaag A, Yasuda T, Gao HZ, Saheki T, Bachmann C, Elpeleg O. Infantile citrullinemia caused by citrin deficiency with increased dibasic amino acids. Mol Genet Metab. 2002;77:202 - 8. [PubMed: 12409267]
- Bijarnia-Mahay S, Häberle J, R, Fenacht VR, Shigematsu Y, Saxena R, Verma IC. Citrin deficiency: A treatable cause of acute psychosis in adults. Neurol India. 2015;63:220 - 2. [PubMed: 25947987]
- Chen R, Wang XH, Fu HY, Zhang SR, Abudouxikuer K, Saheki T, Wang JS. Different regional distribution of SLC25A13 mutations in Chinese patients with neonatal intrahepatic cholestasis. World J Gastroenterol. 2013;19:4545 - 51. [PMC free article: PMC3725380] [PubMed: 23901231]
- del Arco A, Satrústegui J. Molecular cloning of Aralar, a new member of the mitochondrial carrier superfamily that binds calcium and is present in human muscle and brain. J Biol Chem. 1998;273:23327 - 34. [PubMed: 9722566]
- Dimmock D, Kobayashi K, Iijima M, Tabata A, Wong LJ, Saheki T, Lee B, Scaglia F. Citrin deficiency: a novel cause of failure to thrive that responds to a high protein, low carbohydrate diet. Pediatrics. 2007;119:e773 - 7. [PubMed: 17332192]
- Dimmock D, Maranda B, Dionisi-Vici C, Wang J, Kleppe S, Fiermonte G, Bai R, Hainline B, Hamosh A, O'Brien WE, Scaglia F, Wong LJ. Citrin deficiency, a perplexing global disorder. Mol Genet Metab. 2009;96:44 - 9. [PubMed: 19036621]
- Feillet F, Merten M, Battaglia-Hsu SF, Rabier D, Kobayashi K, Straczek J, Brivet M, Favre E, Guéant JL. Evidence of cataplerosis in a patient with neonatal classical galactosemia presenting as citrin deficiency. J Hepatol. 2008;48:517 - 22. [PubMed: 18207281]
- Fiermonte G, Soon D, Chaudhuri A, Paradies E, Lee PJ, Krywawych S, Palmieri F, Lachmann RH. An adult with type 2 citrullinemia presenting in Europe. N Engl J Med. 2008;358:1408 - 9. [PubMed: 18367750]
- Fu HY, Zhang SR, Wang XH, Saheki T, Kobayashi K, Wang JS. The mutation spectrum of the SLC25A13 gene in Chinese infants with intrahepatic cholestasis and aminoacidemia. J Gastroenterol. 2011;46:510 - 8. [PubMed: 20927635]
- Fukumoto K, Sumida Y, Yoshida N, Sakai K, Kanemasa K, Itoh Y, Mitsufuji S, Kataoka K, Okanoue T. A case of adult-onset type II citrullinemia having a liver histology of nonalcoholic steatohepatitis (NASH). Nippon Shokakibyo Gakkai Zasshi. 2008;105:244 - 51. [PubMed: 18250596]
- Gao HZ, Kobayashi K, Tabata A, Tsuge H, Iijima M, Yasuda T, Kalkanoglu HS, Dursun A, Tokatli A, Coskun T, Trefz FK, Skladal D, Mandel H, Seidel J, Kodama S, Shirane S, Ichida T, Makino S, Yoshino M, Kang JH, Mizuguchi M, Barshop BA, Fuchinoue S, Seneca S, Zeesman S, Knerr I, Rodes M, Wasant P, Yoshida I, De Meirleir L, Abdul Jalil M, Begum L, Horiuchi M, Katunuma N, Nakagawa S, Saheki T. Identification of 16 novel mutations in the argininosuccinate synthetase gene and genotype-phenotype correlation in 38 classical citrullinemia patients. Hum Mutat. 2003;22:24 - 34. [PubMed: 12815590]
- Hachisu M, Oda Y, Goto M, Kobayashi K, Saheki T, Ohura T, Noma S, Kitanaka S. Citrin deficiency presenting with ketotic hypoglycaemia and hepatomegaly in childhood. Eur J Pediatr. 2005;164:109 - 10. [PubMed: 15592876]
- Hagiwara N, Sekijima Y, Takei Y, Ikeda S, Kawasaki S, Kobayashi K, Saheki T. Hepatocellular carcinoma in a case of adult-onset type II citrullinemia. Intern Med. 2003;42:978 - 82. [PubMed: 14606711]
- Hayasaka K, Numakura C, Toyota K, Kakizaki S, Watanabe H, Haga H, Takahashi H, Takahashi Y, Kaneko M, Yamakawa M, Nunoi H, Kato T, Ueno Y, Mori M. Medium-chain triglyceride supplementation under a low-carbohydrate formula is a promising therapy for adult-onset type II citrullinemia. Mol Genet Metab Rep. 2014;1:42 - 50. [PMC free article: PMC5121258] [PubMed: 27896073]
- Hayasaka K, Numakura C, Toyota K, Kimura T. Treatment with lactose (galactose)-restricted and medium-chain triglyceride-supplemented formula for neonatal intrahepatic cholestasis caused by citrin deficiency. JIMD Rep. 2012;2:37 - 44. [PMC free article: PMC3509838] [PubMed: 23430852]
- Hirai I, Kimura W, Suto K, Fzjimoto H, Watanabe T, Fuse A, Kobayashi K, Iijima M, Saheki T, Nakatsuka T, Sugawara Y, Makuuchi M. Living donor liver transplantation for type II citrullinemia from a heterozygous donor. Hepatogastroenterology. 2008;55:2211 - 6. [PubMed: 19260507]
- Hutchin T, Preece MA, Kobayashi K, Saheki T, Brown R, Kelly DA, McKiernan PJ, Green A, Baumann U. Neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD) in a European patient. J Inherit Metab Dis. 2006;29 Suppl 1:112.
- Ikeda S, Kawa S, Takei Y, Yamamoto K, Shimojo H, Tabata K, Kobayashi K, Saheki T. Chronic pancreatitis associated with adult-onset type II citrullinemia: clinical and pathologic findings. Ann Intern Med. 2004;141:W109-10. [PubMed: 15466765]
- Ikeda S, Yazaki M, Takei Y, Ikegami T, Hashikura Y, Kawasaki S, Iwai M, Kobayashi K, Saheki T. Type II (adult onset) citrullinaemia: clinical pictures and the therapeutic effect of liver transplantation. J Neurol Neurosurg Psychiatry. 2001;71:663 - 70. [PMC free article: PMC1737600] [PubMed: 11606680]
- Imamura Y, Kobayashi K, Shibatou T, Aburada S, Tahara K, Kubozono O, Saheki T. Effectiveness of carbohydrate-restricted diet and arginine granules therapy for adult-onset type II citrullinemia: a case report of siblings showing homozygous SLC25A13 mutation with and without the disease. Hepatol Res. 2003;26:68 - 72. [PubMed: 12787807]
- Kasahara M, Ohwada S, Takeichi T, Kaneko H, Tomomasa T, Morikawa A, Yonemura K, Asonuma K, Tanaka K, Kobayashi K, Saheki T, Takeyoshi I, Morishita Y. Living-related liver transplantation for type II citrullinemia using a graft from heterozygote donor. Transplantation. 2001;71:157 - 9. [PubMed: 11211185]
- Ko JM, Kim GH, Kim JH, Kim JY, Choi JH, Ushikai M, Saheki T, Kobayashi K, Yoo HW. Six cases of citrin deficiency in Korea. Int J Mol Med. 2007a;20:809 - 15. [PubMed: 17982687]
- Ko JS, Song JH, Park SS, Seo JK. Neonatal intrahepatic cholestasis caused by citrin deficiency in Korean infants. J Korean Med Sci. 2007b;22:952 - 6. [PMC free article: PMC2694627] [PubMed: 18162705]
- Kobayashi K, Horiuchi M, Saheki T. Pancreatic secretory trypsin inhibitor as a diagnostic marker for adult-onset type II citrullinemia. Hepatology. 1997;25:1160 - 5. [PubMed: 9141434]
- Kobayashi K, Iijima M, Ushikai M, Ikeda S, Saheki T. Citrin deficiency. J Jpn Pediatr Soc. 2006;110:1047 - 59.
- Kobayashi K, Iijima M, Yasuda T, Sinasac DS, Yamaguchi N, Tsui L-C, Scherer SW, Saheki T. Type II citrullinemia (citrin deficiency): a mysterious disease caused by a defect of calcium-binding mitochondrial carrier protein. In: Pochet R, ed. Calcium: The Molecular Basis of Calcium Action in Biology and Medicine. Dordrecht, Netherlands: Kluwer; 2000:565-87.
- Kobayashi K, Saheki T. Molecular basis of citrin deficiency. Seikagaku. 2004;76:1543 - 59. [PubMed: 15675368]
- Kobayashi K, Saheki T. Aspartate glutamate carrier (citrin) deficiency. In: Broer S, Wagner CS, eds. Membrane Transporter Diseases. New York, NY: Kluwer Academic/Plenum Publishers; 2003:147-60.
- Kobayashi K, Sinasac DS, Iijima M, Boright AP, Begum L, Lee JR, Yasuda T, Ikeda S, Hirano R, Terazono H, Crackower MA, Kondo I, Tsui LC, Scherer SW, Saheki T. The gene mutated in adult-onset type II citrullinaemia encodes a putative mitochondrial carrier protein. Nat Genet. 1999;22:159 - 63. [PubMed: 10369257]
- Komatsu M, Yazaki M, Tanaka N, Sano K, Hashimoto E, Takei Y, Song YZ, Tanaka E, Kiyosawa K, Saheki T, Aoyama T, Kobayashi K. Citrin deficiency as a cause of chronic liver disorder mimicking non-alcoholic fatty liver disease. J Hepatol. 2008;49:810 - 20. [PubMed: 18620775]
- Lee BH, Jin HY, Kim GH, Choi JH, Yoo HW. Nonalcoholic fatty liver disease in 2 siblings with adult-onset type II citrullinemia. J Pediatr Gastroenterol Nutr. 2010;50:682 - 5. [PubMed: 20400906]
- Lee HC, Mak CM, Lam CW, Yuen YP, Chan AO, Shek CC, Siu TS, Lai CK, Ching CK, Siu WK, Chen SP, Law CY, Tai HL, Tam S, Chan AY. Analysis of inborn errors of metabolism: disease spectrum for expanded newborn screening in Hong Kong. Chin Med J (Engl). 2011;124:983 - 9. [PubMed: 21542954]
- Lee NC, Chien YH, Kobayashi K, Saheki T, Chen H-L, Chiu PC, Ni YH, Chang MH, Hwu WL. Time course of acylcarnitine elevation in neonatal intrahepatic cholestasis caused by citrin deficiency. J Inherit Metab Dis. 2006;29:551 - 5. [PubMed: 16736097]
- Lin WX, Zeng HS, Zhang ZH, Mao M, Zheng QQ, Zhao ST, Cheng Y, Chen FP, Wen WR, Song YZ. Molecular diagnosis of pediatric patients with citrin deficiency in China: SLC25A13 mutation spectrum and the geographic distribution. Sci Rep. 2016;6:29732. [PMC free article: PMC4942605] [PubMed: 27405544]
- Lin WX, Zhang ZH, Deng M, Cai XR, Song YZ. Multiple ovarian antral follicles in a preterm infant with neonatal intrahepatic cholestasis caused by citrin deficiency: A clinical, genetic and transcriptional analysis. Gene. 2012;505:269 - 75. [PubMed: 22710133]
- Lu YB, Kobayashi K, Ushikai M, Tabata A, Iijima M, Li MX, Lei L, Kawabe K, Taura S, Yang Y, Liu TT, Chiang SH, Hsiao KJ, Lau YL, Tsui LC, Lee DH, Saheki T. Frequency and distribution in East Asia of 12 mutations identified in the SLC25A13 gene of Japanese patients with citrin deficiency. J Hum Genet. 2005;50:338 - 46. [PubMed: 16059747]
- Luder AS, Tabata A, Iijima M, Kobayashi K, Mandel H. Citrullinaemia type 2 outside East Asia: Israeli experience. J Inherit Metab Dis. 2006;29:59.
- Miyakoshi T, Takahashi T, Kato M, Watanabe M, Ito C. Abnormal citrulline metabolism of Inose-type hepatocerebral disease. Shinkei Kagaku. 1968;7:88 - 91.
- Mutoh K, Kurokawa K, Kobayashi K, Saheki T. Treatment of a citrin-deficient patient at the early stage of adult-onset type II citrullinaemia with arginine and sodium pyruvate. J Inherit Metab Dis. 2008;31 Suppl 2:S343 - 7. [PubMed: 18958581]
- Nagasaka H, Komatsu H, Inui A, Nakacho M, Morioka I, Tsukahara H, Kaji S, Hirayama S, Miida T, Kondou H, Ihara K, Yagi M, Kizaki Z, Bessho K, Kodama T, Iijima K, Saheki T, Yorifuji T, Honda A. Circulating tricarboxylic acid cycle metabolite levels in citrin-deficient children with metabolic adaptation, with and without sodium pyruvate treatment. Mol Genet Metab. 2017;120:207 - 12. [PubMed: 28041819]
- Nagasaka H, Okano Y, Tsukahara H, Shigematsu Y, Momoi T, Yorifuji J, Miida T, Ohura T, Kobayashi K, Saheki T, Hirano K, Takayanagi M, Yorifuji T. Sustaining hypercitrullinemia, hypercholesterolemia and augmented oxidative stress in Japanese children with aspartate/glutamate carrier isoform 2-citrin-deficiency even during the silent period. Mol Genet Metab. 2009;97:21 - 26. [PubMed: 19232506]
- Oh SH, Lee BH, Kim GH, Choi JH, Kim KM, Yoo HW. Biochemical and molecular characteristics of citrin deficiency in Korean children. J Hum Genet. 2017;62:305 - 7. [PubMed: 27829683]
- Ohura T, Abukawa D, Aikawa J, Nakagawa M, Ishizawa S, Tazawa Y, Narisawa K, Iinuma K. Disturbances of amino acid metabolism in infants with idiopathic neonatal hepatitis. J Jpn Pediatr Soc. 1997;101:1522 - 5.
- Ohura T, Kobayashi K, Abukawa D, Tazawa Y, Aikawa J, Sakamoto O, Saheki T, Iinuma K. A novel inborn error of metabolism detected by elevated methionine and/or galactose in newborn screening: neonatal intrahepatic cholestasis caused by citrin deficiency. Eur J Pediatr. 2003;162:317 - 22. [PubMed: 12692712]
- Ohura T, Kobayashi K, Tazawa Y, Abkawa D, Sakamoto O, Tsuchiya S, Saheki T. Clinical pictures of 75 patients with neonatal intrahepatic cholestasis caused by citrin deficiency. J Inherit Metab Dis. 2007;30:139 - 44. [PubMed: 17323144]
- Okano Y, Kobayashi K, Ihara K, Ito T, Yoshino M, Watanabe Y, Kaji S, Ohura T, Nagao M, Noguchi A, Mushiake S, Hohashi N, Hashimoto-Tamaoki T. Fatigue and quality of life in citrin deficiency during adaptation and compensation stage. Mol Genet Metab. 2013;109:9 - 13. [PubMed: 23453692]
- Palmieri L, Pardo B, Lasorsa FM, del Arco A, Kobayashi K, Iijima M, Runswick MJ, Walker JE, Saheki T, Satrustegui J, Palmieri F. Citrin and aralar1 are Ca(2+)-stimulated aspartate/glutamate transporters in mitochondria. EMBO J. 2001;20:5060 - 9. [PMC free article: PMC125626] [PubMed: 11566871]
- Saheki T, Iijima M, Li MX, Kobayashi K, Horiuchi M, Ushikai M, Okumura F, Meng XJ, Inoue I, Tajima A, Moriyama M, Eto K, Kadowaki T, Sinasac DS, Tsui L-C, Tsuji M, Okano A, Kobayashi T. Citrin/mitochondrial glycerol-3-phosphate dehydrogenase double-knockout mice recapitulate features of human citrin deficiency. J Biol Chem. 2007;282:25041 - 52. [PubMed: 17591776]
- Saheki T, Inoue K, Tushima A, Mutoh K, Kobayashi K. Citrin deficiency and current treatment concepts. Mol Genet Metab. 2010;100:S59 - 64. [PubMed: 20233664]
- Saheki T, Kobayashi K. Mitochondrial aspartate glutamate carrier (citrin) deficiency as the cause of adult-onset type II citrullinemia (CTLN2) and idiopathic neonatal hepatitis (NICCD). J Hum Genet. 2002;47:333 - 41. [PubMed: 12111366]
- Saheki T, Kobayashi K. Physiological role of citrin, a liver-type mitochondrial aspartate-glutamate carrier, and pathophysiology of citrin deficiency. Recent Res Devel Life Sci. 2005;3:59 - 73.
- Saheki T, Kobayashi K, Iijima M, Horiuchi M, Begum L, Jalil MA, Li MX, Lu YB, Ushikai M, Tabata A, Moriyama M, Hsiao KJ, Yang Y. Adult-onset type II citrullinemia and idiopathic neonatal hepatitis caused by citrin deficiency: involvement of the aspartate glutamate carrier for urea synthesis and maintenance of the urea cycle. Mol Genet Metab. 2004;81 Suppl 1:S20 - 6. [PubMed: 15050970]
- Saheki T, Kobayashi K, Iijima M, Li MX, Horiuchi M, Tabata A, Lu YB, Ushikai M, Terashi M, Moriyama M. Pathophysiology of citrin deficiency. In: Haussinger D, Kircheis G, Schliess F, eds. Hepatic Encephalopathy and Nitrogen Metabolism. Dordrecht, Netherlands: Springer; 2006:320-8.
- Saheki T, Kobayashi K, Terashi M, Ohura T, Yanagawa Y, Okano Y, Hattori T, Fujimoto H, Mutoh K, Kizaki Z, Inui A. Reduced carbohydrate intake in citrin-deficient subjects. J Inherit Metab Dis. 2008;31:386 - 94. [PubMed: 18415701]
- Saheki T, Nakano K, Kobayashi K, Imamura Y, Itakura Y, Sase M, Hagihara S, Matuo S. Analysis of the enzyme abnormality in eight cases of neonatal and infantile citrullinaemia in Japan. J Inherit Metab Dis. 1985;8:155 - 6. [PubMed: 3939592]
- Saheki T, Ueda A, Hosoya M, Kusumi K, Takada S, Tsuda M, Katsunuma T. Qualitative and quantitative abnormalities of argininosuccinate synthetase in citrullinemia. Clin Chim Acta. 1981;109:325 - 35. [PubMed: 6784969]
- Shigematsu Y, Hirano S, Hata I, Tanaka Y, Sudo M, Sakura N, Tajima T, Yamaguchi S. Newborn mass screening and selective screening using electrospray tandem mass spectrometry in Japan. J Chromatogr B Analyt Technol Biomed Life Sci. 2002;776:39 - 48. [PubMed: 12127323]
- Shiohama N, Sugita Y, Imamura N, Sato T, Mizuno Y. Type II citrullinemia triggered by acetaminophen. No To Shinkei. 1993;45:865 - 70. [PubMed: 8217412]
- Sinasac DS, Crackower MA, Lee JR, Kobayashi K, Saheki T, Scherer SW, Tsui LC. Genomic structure of the adult-onset type II citrullinemia gene, SLC25A13, and cloning and expression of its mouse homologue. Genomics. 1999;62:289 - 92. [PubMed: 10610724]
- Soeda J, Yazaki M, Nakata T, Miwa S, Ikeda S, Hosoda W, Iijima M, Kobayashi K, Saheki T, Kojiro M, Miyagawa S. Primary liver carcinoma exhibiting dual hepatocellular-biliary epithelial differentiations associated with citrin deficiency: a case report. J Clin Gastroenterol. 2008;42:855 - 60. [PubMed: 18385606]
- Song YZ, Deng M, Chen FP, Wen F, Guo L, Cao SL, Gong J, Xu H, Jiang GY, Zhong L, Kobayashi K, Saheki T, Wang ZN. Genotypic and phenotypic features of citrin deficiency: Five-year experience in a Chinese pediatric center. Int J Mol Med. 2011;28:33 - 40. [PubMed: 21424115]
- Song YZ, Guo L, Yang YL, Han LS, Kobayashi K, Saheki T. Failure to thrive and dyslipidemia caused by citrin deficiency: a novel clinical phenotype. Zhongguo Dang Dai Er Ke Za Zhi. 2009a;11:328 - 32. [PubMed: 19470249]
- Song YZ, Hao H, Ushikai M, Liu GS, Xiao X, Saheki T, Kobayashi K, Wang ZN. A difficult and complicated case study: neonatal intrahepatic cholestasis caused by citrin deficiency. Zhongguo Dang Dai Er Ke Za Zhi. 2006;8:125 - 8. [PubMed: 16613706]
- Song YZ, Li BX, Chen FP, Liu SR, Sheng JS, Ushikai M, Zhang CH, Zhang T, Wang ZN, Kobayashi K, Saheki T, Zheng XY. Neonatal intrahepatic cholestasis caused by citrin deficiency: clinical and laboratory investigation of 13 subjects in mainland of China. Dig Liver Dis. 2009b;41:683 - 9. [PubMed: 19185551]
- Song YZ, Sheng JS, Ushikai M, Hwu WL, Zhang CH, Kobayashi K. Identification and diagnosis of three novel mutations in SLC25A13 gene of neonatal intrahepatic cholestasis caused by citrin deficiency. Zhonghua Er Ke Za Zhi. 2008;46:411 - 5. [PubMed: 19099775]
- Song YZ, Wen F, Chen FP, Kobayashi K, Saheki T. Neonatal intrahepatic cholestasis caused by citrin deficiency: efficacy of therapeutic formulas and update of clinical outcomes. Jpn J Inherit Metab Dis. 2010;26:57 - 69.
- Song YZ, Zhang ZH, Lin WX, Zhao XJ, Deng M, Ma YL, Guo L, Chen FP, Long XL, He XL, Sunada Y, Soneda S, Nakatomi A, Dateki S, Ngu LH, Kobayashi K, Saheki T. SLC25A13 gene analysis in citrin deficiency: sixteen novel mutations in Asian patients, and the mutation distribution in a large pediatric cohort in China. PLoS One. 2013;8:e74544. [PMC free article: PMC3777997] [PubMed: 24069319]
- Tabata A, Sheng J-S, Ushikai M, Song Y-Z, Gao H-Z, Lu Y-B, Okumura F, Iijima M, Mutoh K, Kishida S, Saheki T, Kobayashi K. Identification of 13 novel mutations including a retrotransposal insertion in SLC25A13 gene and frequency of 30 mutations found in patients with citrin deficiency. J Hum Genet. 2008;53:534 - 45. [PubMed: 18392553]
- Takagi H, Hagiwara S, Hashizume H, Kanda D, Sato K, Sohara N, Kakizaki S, Takahashi H, Mori M, Kaneko H, Ohwada S, Ushikai M, Kobayashi K, Saheki T. Adult onset type II citrullinemia as a cause of non-alcoholic steatohepatitis. J Hepatol. 2006;44:236 - 9. [PubMed: 16278034]
- Takahashi H, Kagawa T, Kobayashi K, Hirabayashi H, Yui M, Begum L, Mine T, Takagi S, Saheki T, Shinohara Y. A case of adult-onset type II citrullinemia - deterioration of clinical course after infusion of hyperosmotic and high sugar solutions. Med Sci Monit. 2006;12:CS13 - 5. [PubMed: 16449956]
- Takahashi Y, Koyama S, Tanaka H, Arawaka S, Wada M, Kawanami T, Haga H, Watanabe H, Toyota K, Numakura C, Hayasaka K, Kato T. An elderly Japanese patient with adult-onset type II citrullinemia with a novel D493G mutation in the SLC25A13 gene. Intern Med. 2012;51:2131 - 4. [PubMed: 22892490]
- Takaya J, Kobayashi K, Ohashi A, Ushikai M, Tabata A, Fujimoto S, Yamato F, Saheki T, Kobayashi Y. Variant clinical courses of 2 patients with neonatal intrahepatic cholestasis who have a novel mutation of SLC25A13. Metabolism. 2005;54:1615 - 9. [PubMed: 16311094]
- Takeuchi S, Yazaki M, Yamada S, Fukuyama T, Inui A, Iwasaki Y, Ikeda S (2015) An adolescent case of citrin deficiency with severe anorexia mimicking anorexia nervosa. Pediatrics136:e530-4. [PubMed: 26195537]
- Tamakawa S, Nakamura H, Katano T, Yoshizawa M, Ohtake K, Kubota T. Hyperalimentation therapy produces a comatose state in a patient with citrullinemia. J Jpn Soc Intensive Care Med. 1994;1:37 - 41.
- Tamamori A, Fujimoto A, Okano Y, Kobayashi K, Saheki T, Tagami Y, Takei H, Shigematsu Y, Hata I, Ozaki H, Tokuhara D, Nishimura Y, Yorifuji T, Igarashi N, Ohura T, Shimizu T, Inui K, Sakai N, Abukawa D, Miyakawa T, Matsumori M, Ban K, Kaneko H, Yamano T. Effects of citrin deficiency in the perinatal period: feasibility of newborn mass screening for citrin deficiency. Pediatr Res. 2004;56:608 - 14. [PubMed: 15295082]
- Tamamori A, Okano Y, Ozaki H, Fujimoto A, Kajiwara M, Fukuda K, Kobayashi K, Saheki T, Tagami Y, Yamano T. Neonatal intrahepatic cholestasis caused by citrin deficiency: severe hepatic dysfunction in an infant requiring liver transplantation. Eur J Pediatr. 2002;161:609 - 13. [PubMed: 12424587]
- Tanaka T, Nagao M, Tsutsumi H. Application of mutation analysis for the previously uncertain cases of adult-onset type II citrullinemia (CTLN2) and their clinical profiles. Tohoku J Exp Med. 2002;198:89 - 97. [PubMed: 12512993]
- Tazawa Y, Abukawa D, Sakamoto O, Nagata I, Murakami J, Iizuka T, Okamoto M, Kimura A, Kurosawa T, Iinuma K, Kobayashi K, Saheki T, Ohura T. A possible mechanism of neonatal intrahepatic cholestasis caused by citrin deficiency. Hepatol Res. 2005;31:168 - 71. [PubMed: 15777702]
- Tokuhara D, Iijima M, Tamamori A, Ohura T, Takaya J, Maisawa S, Kobayashi K, Saheki T, Yamano T, Okano Y. Novel diagnostic approach to citrin deficiency: Analysis of citrin protein in lymphocytes. Mol Genet Metab. 2007;90:30 - 6. [PubMed: 17092749]
- Tomomasa T, Kobayashi K, Kaneko H, Shimura H, Fukusato T, Tabata M, Inoue Y, Ohwada S, Kasahara M, Morishita Y, Kimura M, Saheki T, Morikawa A. Possible clinical and histologic manifestations of adult-onset type II citrullinemia in early infancy. J Pediatr. 2001;138:741 - 3. [PubMed: 11343053]
- Tong F, Yang JB, Huang XL, Zhou XL, Yang RL. A case of neonatal intrahepatic cholestasis caused by citrin deficiency complicated with congenital biliary atresia. Zhonghua Er Ke Za Zhi. 2013;51:863 - 5. [PubMed: 24484564]
- Tsai C-W, Yang C-C, Chen H-L, Hwu W-L, Wu M-Z, Liu K-L, Wu M-S. Homozygous SLC25A13 mutation in a Taiwanese patient with adult-onset citrullinemia complicated with steatosis and hepatocellular carcinoma. J Formos Med Assoc. 2006;105:852 - 6. [PubMed: 17000460]
- Tsuboi Y, Fujino Y, Kobayashi K, Saheki T, Yamada T. High serum pancreatic secretory trypsin inhibitor before onset of type II citrullinemia. Neurology. 2001;57:933. [PubMed: 11552040]
- Wang H, Shu S, Chen C, Huang Z, Wang D. Novel mutations in the SLC25A13 gene in a patient with NICCD and severe manifestations. J Pediatr Endocrinol Metab. 2015;28:471 - 5. [PubMed: 25381944]
- Wen PQ, Wang GB, Chen ZL, Liu XH, Cui D, Shang Y, Li CR. Clinical investigation and mutation analysis of a child with citrin deficiency complicated with purpura, convulsive seizures and methioninemia. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2013;30:649 - 53. [PubMed: 24327139]
- Wong LJ, Dimmock D, Geraghty MT, Quan R, Lichter-Konecki U, Wang J, Brundage EK, Scaglia F, Chinault AC. Utility of oligonucleotide array-based comparative genomic hybridization for detection of target gene deletions. Clin Chem. 2008;54:1141 - 8. [PubMed: 18487280]
- Xing YZ, Qiu WJ, Ye J, Han LS, Xu SS, Zhang HW, Gao XL, Wang Y, Gu XF. Studies on the clinical manifestation and SLC25A13 gene mutation of Chinese patients with neonatal intrahepatic cholestasis caused by citrin deficiency. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2010;27:180 - 5. [PubMed: 20376801]
- Xiong XL, Yan SQ, Ding Y, Zhou LS, Chen P. Clinical research of neonatal intrahepatic cholestasis caused by citrin deficiency in Hubei Province. Ch J Appl Clin Pediatr. 2015;14:1064 - 8. ZHao DC.
- Yamaguchi N, Kobayashi K, Yasuda T, Nishi I, Iijima M, Nakagawa M, Osame M, Kondo I, Saheki T. Screening of SLC25A13 mutations in early and late onset patients with citrin deficiency and in the Japanese population: Identification of two novel mutations and establishment of multiple DNA diagnosis methods for nine mutations. Hum Mutat. 2002;19:122 - 30. [PubMed: 11793471]
- Yasuda T, Yamaguchi N, Kobayashi K, Nishi I, Horinouchi H, Jalil MA, Li MX, Ushikai M, Iijima M, Kondo I, Saheki T. Identification of two novel mutations in the SLC25A13 gene and detection of seven mutations in 102 patients with adult-onset type II citrullinemia. Hum Genet. 2000;107:537 - 45. [PubMed: 11153906]
- Yazaki M, Hashikura Y, Takei Y, Ikegami T, Miyagawa S, Yamamoto K, Tokuda T, Kobayashi K, Saheki T, Ikeda S. Feasibility of auxiliary partial orthotopic liver transplantation from living donors for patients with adult-onset type II citrullinemia. Liver Transpl. 2004;10:550 - 4. [PubMed: 15048800]
- Yazaki M, Ikeda S, Kobayashi K, Saheki T. Therapeutic approaches for patients with adult-onset type II citrullinemia (CTLN2): effectiveness of treatment with low-carbohydrate diet and sodium pyruvate. Rinsho Shinkeigaku. 2010;50:844 - 7. [PubMed: 21921468]
- Yazaki M, Takei Y, Kobayashi K, Saheki T, Ikeda S. Risk of worsened encephalopathy after intravenous glycerol therapy in patients with adult-onset type II citrullinemia (CTLN2). Intern Med. 2005;44:188 - 95. [PubMed: 15805705]
- Yeh JN, Jeng YM, Chen HL, Ni YH, Hwu WL, Chang MH. Hepatic steatosis and neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD) in Taiwanese infants. J Pediatr. 2006;148:642 - 6. [PubMed: 16737877]
- Zeng HS, Lin WX, Zhao ST, Zhang ZH, Yang HW, Chen FP, Song YZ, Yin ZN. SLC25A13c DNA cloning analysis using peripheral blood lymphocytes facilitates the identification of a large deletion mutation: Molecular diagnosis of an infant with neonatal intrahepatic cholestasis caused by citrin deficiency. Mol Med Rep. 2016;14:5189 - 94. [PubMed: 27779681]
- Zeng HS, Zhao ST, Deng M, Zhang ZH, Cai XR, Chen FP, Song YZ. Inspissated bile syndrome in an infant with citrin deficiency and congenital anomalies of the biliary tract and esophagus: Identification and pathogenicity analysis of a novel SLC25A13 mutation with incomplete penetrance. Int J Mol Med. 2014;34:1241 - 8. [PMC free article: PMC4199400] [PubMed: 25216257]
- Zhang ZH, Lin WX, Deng M, Zhao ST, Zeng HS, Chen FP, Song YZ. Clinical, molecular and functional investigation on an infant with neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD). PLoS One. 2014a;9:e89267. [PMC free article: PMC3931723] [PubMed: 24586645]
- Zhang ZH, Lin WX, Zheng QQ, Guo L, Zhou QH, Song YZ. Molecular diagnosis of citrin deficiency in an infant with intrahepatic cholestasis: identification of a 21.7kb gross deletion that completely silences the transcriptional and translational expression of the affected SLC25A13 allele. Oncotarget. 2017 [PMC free article: PMC5675625] [PubMed: 29152073]
- Zhang ZH, Yang ZG, Chen FP, Kikuchi A, Liu ZH, Kuang LZ, Li WM, Song YZ, Kure S, Saheki S. Screening for five prevalent mutations of SLC25A13 gene in Guangdong, China: A molecular epidemiological survey of citrin deficiency. Tohoku J Exp Med. 2014b;233:275 - 81. [PubMed: 25110155]
- Zheng QQ, Zhang ZH, Zeng HS, Lin WX, Yang HW, Yin ZN, Song YZ. Identification of a large SLC25A13 deletion via sophisticated molecular analyses using peripheral blood lymphocytes in an infant with Neonatal Intrahepatic Cholestasis caused by Citrin Deficiency (NICCD): A clinical and molecular study. Biomed Res Int. 2016;2016:4124263. [PMC free article: PMC4835617] [PubMed: 27127784]
章节注释
作者注
The first author of this review, Keiko Kobayashi, PhD, died of colon cancer on December 21, 2010. The scientific community has lost a great scientist, teacher, and friend.
Keiko Kobayashi is recognized internationally as a pioneer in citrin deficiency research. As an investigator in the research group of Professor Takeyori Saheki (Department of Molecular Metabolism and Biochemical Genetics, Kagoshima University, Japan), in 1999 she cloned the causative 基因 SLC25A13 and designated the term "citrin" for citrin deficiency. Kobayashi also played essential roles in the discovery and designation of NICCD and FTTDCD, two early-onset forms of citrin deficiency. As an outstanding molecular geneticist, she identified more than 50 pathogenic variants in SLC25A13 and diagnosed more than 500 citrin-deficient patients worldwide(Japan, Korea, China, Vietnam, Malaysia, Israel, Palestine, Australia, Czech, France, Britain, and the US). She also worked tirelessly to educate the medical community about citrin deficiency, thus improving the care and prognosis of 受累的 patients worldwide. Less than a month before her death, Dr Kobayashi delivered a lecture on citrin deficiency to the 9th Asia-Pacific Conference on Human Genetics.
Keiko Kobayashi, the "mother of citrin deficiency," will be remembered and sorely missed by her friends, students, colleagues, and the citrin-deficient patients whom she diagnosed.
Acknowledgments鸣谢
This research was supported in part by Grants-in-Aid for Scientific Research (Nos. 16390100, 19390096 and 19591230) and for Asia-Africa Scientific Platform Program (AASPP) from the Japan Society for the Promotion of Science (JSPS), by a Grant for Child Health and Development (17-2) from the Ministry of Health, Labour and Welfare in Japan, by a Grant for Research for Promoting Technological Seeds from the Japan Science and Technology Agency and by the Projects 81070279,81270957 and 81570793 supported by the National Natural Science Foundation (NSFC) of China.
作者记录
Keiko Kobayashi, PhD; Kagoshima University Graduate School of Medical and Dental Sciences, Kagoshima, Japan (2004-2010)
Takeyori Saheki, MD, PhD (2004-present)
Yuan-Zong Song, MD, PhD (2012-present)
修订记录
- 10 August 2017 (ha) Comprehensive update posted live
- 31 July 2014 (me) Comprehensive update posted live
- 5 January 2012 (me) Comprehensive update posted live
- 1 July 2008 (me) Comprehensive update posted live
- 28 December 2006 (kk) Revision: 序列分析 for SLC25A13 clinically available
- 16 September 2005 (me) Review posted live
- 5 November 2004 (kk) Original submission