
总论
临床特点
粘多糖贮积症IVA的表型谱(MPS IVA)是连续的,从严重的迅速进展的早发型到缓慢渐进的迟发型。MPS IVA患儿出生时通常没有特殊的临床表现。 严重的类型在1到3岁之间发病,经常表现为脊柱侧后弯曲畸形,膝外翻,和鸡胸;缓慢渐进的类型直到童年或青春期可能都不会有明显的临床表现。这一类型往往首先表现为髋关节的问题(疼痛、僵硬和Legg-Perthes病)。渐进性骨关节受累导致身材矮小,最终致残。其他器官系统受累的几率很高,包括呼吸困难,阻塞性睡眠呼吸暂停、瓣膜性心脏病、听力障碍、视力障碍角膜混浊、牙齿异常、肝肿大。脊髓压迫症是一种常见的并发症,导致神经功能缺损。MPS IVA患儿在疾病开始时有正常的智力水平。
诊断/测试
通过N-乙酰氨基半乳糖6-硫酸酯酶(GALNS)活性分析, 或者用分子遗传学手段检测到GALNS基因的双等位突变。
患者管理
对症治疗:使用酶替代疗法(elosulfase alfa,或vimizim™)。但这种治疗对于患儿的骨骼和骨骼外表型的长期效果仍不清楚。MPS IVA的治疗与评价最好由多学科的专家联合实行,并由具有处理复杂临床情况经验的内科医师进行协助。理疗师、物理治疗师、职业治疗师有助于改进运动性和自主性。心理学支持有助于提高应对技能和生活质量;教育专家可以为患者优化个人学习环境。手术治疗的常见指征包括下肢力线不良,髋关节半脱位和/或髋关节疼痛,高位颈椎不稳,和/或渐进的胸腰椎侧后弯曲畸形。上肢治疗可能包括稳定的外部手腕夹板或部分或完整的腕关节融合。心脏瓣膜受累可能需要植入生物或人工瓣膜。上气道阻塞和阻塞性睡眠呼吸暂停可切除扁桃体和腺样体。弥漫性狭窄气道可能需要正压通气和/或气管切开。因治疗角膜混浊而进行的角膜移植术的术后效果因人而异。听力损失最初使用通风管,后来使用助听器。
预防并发症:要预判由脊柱畸形所引起的术前术后麻醉问题及呼吸管理上的困难。全部受影响个人应该接受流感和肺炎球菌疫苗接种以及常规免疫接种。对于那些使用人工心脏瓣膜,心脏瓣膜修复材料,或有感染性心内膜炎既往史的患者,建议进行细菌性心内膜炎的预防。
随访与监护:对于那些进行酶替代治疗的患者,至少每六个月进行一次体检;年度评估生活质量参数与肺功能检查。对所有患者,进行年度耐力测试来评估心血管,肺,肌肉,和神经系统的功能状态;上下肢功能和力线的年度评估;异型增生/半脱位和胸腰椎后凸畸形的评估;每六个月进行神经系统检查和颈椎放射学检查以评估脊髓受压情况;推荐一到三年进行一次全脊柱MRI。根据病程,每一到三年进行心率、心电图、超声心动图检查。年度评估阻塞性睡眠呼吸暂停患者的肺功能;采用MPS IVA患者专用的生长发育图标来评估患者的营养状况。每次随诊时进行视觉和眼睛检查,没六到12个月进行一次牙科检查,每年进行听力评估。
需要避免的身体状况和药物:体重过度增加;β受体阻滞剂。
遗传咨询 MPS IVA遵循常染色体隐性遗传的方式。理论上,患者的兄弟姐妹有25%的机会患病,无症状突变携带者的几率是50%,完全正常的几率是25%。如果致病突变已确定,可以对高风险家庭成员进行携带者检测,或者对高风险妊娠进行产前检查。
诊断
临床线索
当一个人在病史,体格检查、骨骼X线片,眼科检查及实验室检查有以下表现时,应怀疑粘多糖贮积症IVA(MPS IVA)。
- No distinctive clinical findings at birth
- History of adenoidectomy, tonsillectomy, hernia repair, ear ventilation tubes (general findings for all MPS disorders)
- History of cervical spine decompression and/or fusion or a history of surgery for limb alignments (unique to MPS IVA among all MPS disorders)
- Respiratory compromise (sleep apnea, endurance limitations, snoring)
- Cardiac valve abnormalities
- Dental abnormalities
Physical examination. In severe MPS IVA the following findings are usually observed between ages one and three years; in slowly progressive MPS IVA the following findings may not become evident until as late as the second decade of life:
- Marked disproportionate short stature with short trunk and normal limbs (arm span exceeds height)
- Gibbus (short-segment structural thoracolumbar kyphosis resulting in sharp angulation of the back), kyphosis, and scoliosis
- Genu valgum (knock-knee) (Figure 5)
- Hypermobile joints
- Waddling gait with frequent falls
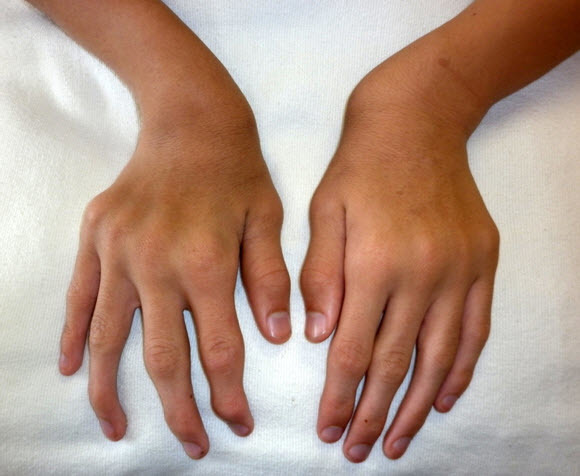
Figure 1.
Ulnar deviation of both wrists and joint enlargement in a male age 15 years with MPS IVA
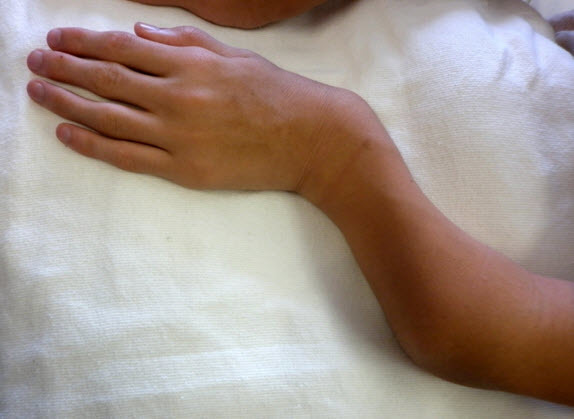
Figure 2.
Shortened forearm and ulnar deviation of the wrist in a male age 15 years with MPS IVA
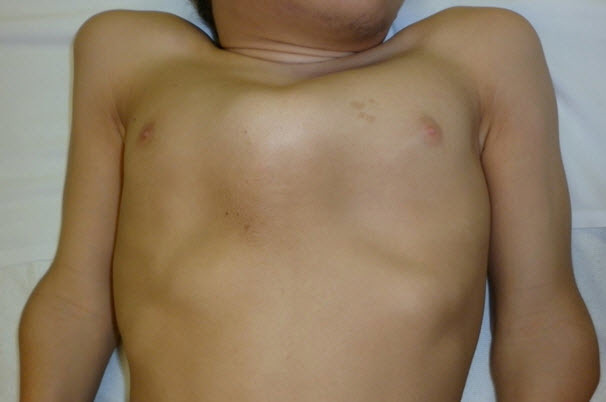
Figure 3.
Pectus anomaly and short neck in a male age 15 years with MPS IVA
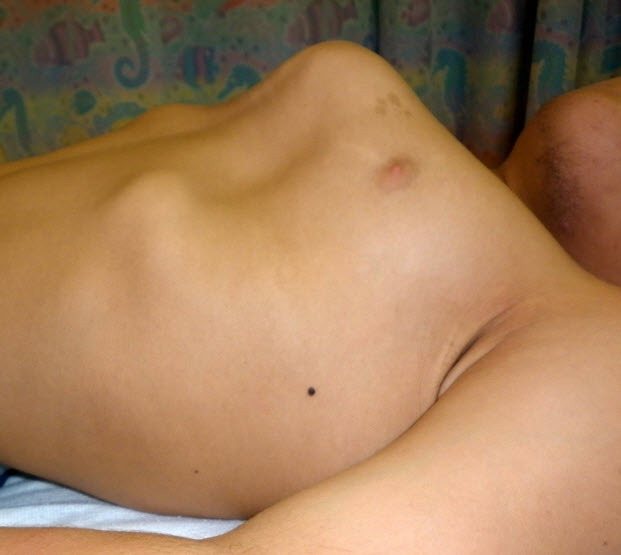
Figure 4.
Lateral view of chest showing severe pectus carinatum in a male age 15 years with MPS IVA
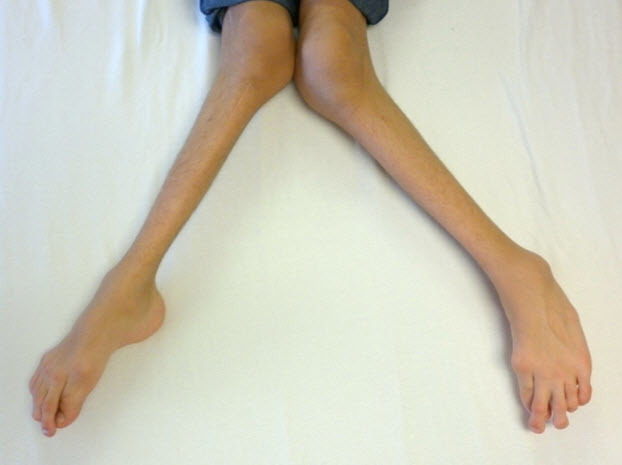
Figure 5.
Severe genu valgum (knock-knee) in a male age 15 years with MPS IVA
Skeletal radiographs
- Odontoid hypoplasia with subsequent cervical instability (Figure 6)
- Kyphosis (curving of the spine that causes a bowing or rounding of the back, which leads to a hunchback or slouching posture) (Figure 7)
- Gibbus (structural kyphosis) with wedging of one or more adjacent vertebrae) (Figure 7)
Note: The radiographic abnormalities of the lumbar spine can be detected at birth in infants with rapidly progressive disease [Tomatsu et al 2011]. - Scoliosis
- Pectus carinatum or (less frequently) excavatum
- Short ulnas, ulnar deviation of the radial epiphysis, and delayed bone maturation
- Short metacarpals with the proximal ends of the second to fifth metacarpals rounded or pointed [White et al 2014]
- Flared iliac wings, flattening of femoral epiphyses (Figure 8), and coxa valga
Note: Skeletal abnormalities are observed before physical abnormalities [Montaño et al 2007, Tomatsu et al 2011].
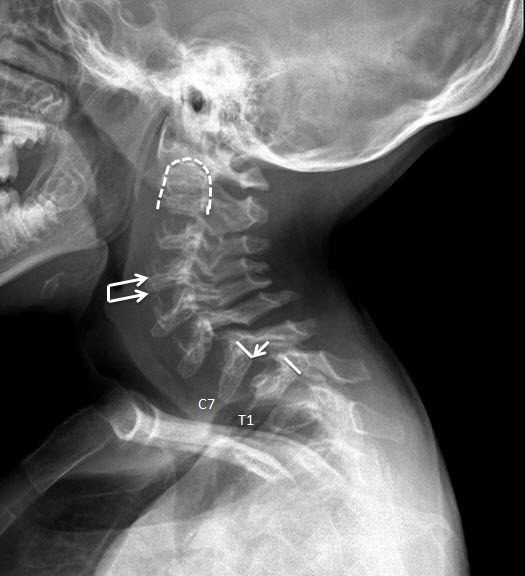
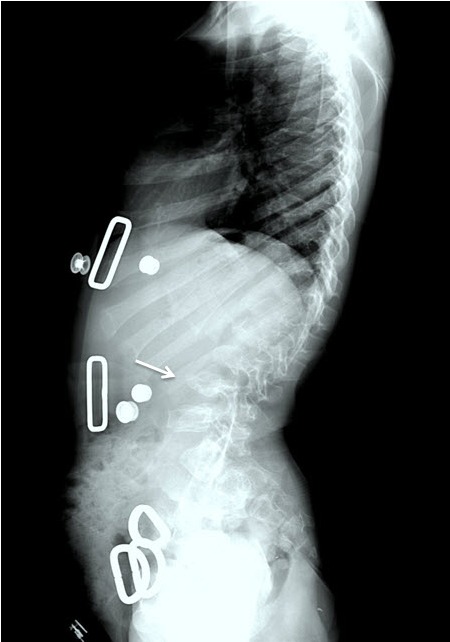
Figure 7.
Lateral spine x-ray of a female age eight years with MPS IVA. Note platyspondyly (flattened vertebrae) with anterior beaking (arrow).
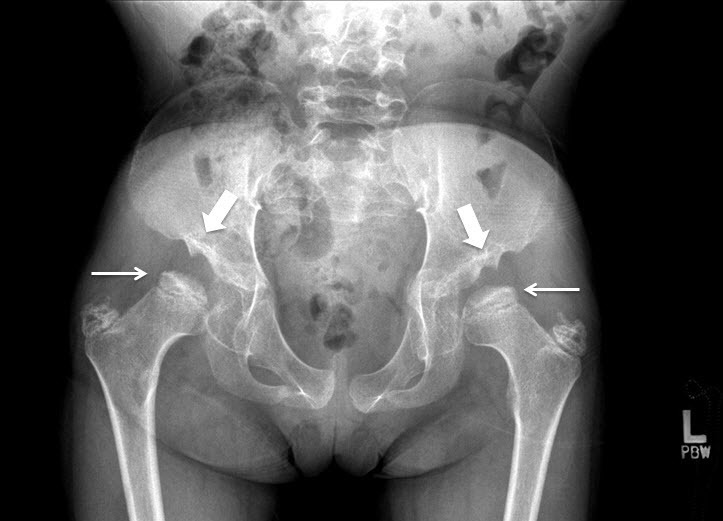
Figure 8.
Hip x-ray of a female age eight years with MPS IVA. Note bilateral irregular flattening of the capital femoral epiphyses (thin arrows), irregular dysplastic acetabuli with lateral joint subluxation (thick arrows).
Ophthalmologic examination. Visual impairment secondary to corneal clouding, astigmatism, and/or retinopathy
Suggestive laboratory findings
- Qualitative urine glycosaminoglycan (GAG) analysis, which uses thin layer chromatography or electrophoresis to identify specific types of GAG [Wood et al 2013], demonstrates keratan sulfate and chondroitin 6-sulfate.
Note: The presence of keratan sulfate (on qualitative analysis) with or without abnormal quantitative urine GAGs has been observed in some 受累的 individuals. - Quantitative urine GAG analysis, which measures the total amount of GAG, demonstrates:
- Elevated keratan sulfate (KS), indicating deficiency of either the enzyme N-acetylgalactosamine 6-sulfatase (in MPS IVA) or the enzyme B-galactosidase (in MPS IVB);
Note: Urine KS levels in younger individuals (≤18 years) is higher than in older individuals due to the decrease in cartilage formation in older persons [Harmatz et al 2013]. - Elevated chondroitin 6-sulfate (C6S), indicating deficiency of the enzyme N-acetylglactosamine 6-sulfatase (MPS IVA).
- Urine keratan sulfate analysis is more sensitive than standard dye-based total GAG quantitative analysis and may be used to replace urine total GAGs in the future.
- Both qualitative and quantitative urine GAGs can be normal in some 受累的 individuals. Thus, further enzymatic or molecular evaluation of a child with clinical evidence of MPS IV is warranted even when GAG analysis is normal.
Establishing the Diagnosis
The diagnosis of MPS IVA is established in a 先证者 with: (1) low N-acetylgalactosamine 6-sulfatase (GALNS) enzyme activity in cultured fibroblasts or leukocytes OR (2) 双等位基因的 pathogenic variants in GALNS identified on 分子遗传学检测 (see Table 1).
Molecular testing approaches can include single-基因 testing or use of a 表型靶向检测.
- Single-基因 testing. Sequence analysis of GALNS is performed first followed by gene-targeted deletion/duplication analysis if only one or no 致病性变异 is found.
- A 表型靶向检测 that includes GALNS and other genes of interest (see Differential Diagnosis) may also be considered. Note: (1) The genes included in the panel and the diagnostic 敏感性 of the testing used for each gene vary by laboratory and over time. (2) Some multi-gene panels may include genes not associated with the condition discussed in this GeneReview; thus, clinicians need to determine which multi-gene panel provides the best opportunity to identify the genetic cause of the condition at the most reasonable cost.
Table 1.
Molecular Genetic Testing Used in Mucopolysaccharidosis Type IVA
| Gene 1 | Test Method | Proportion of Probands with Pathogenic Variants 2 Detectable by This Method |
|---|---|---|
| GALNS | Sequence analysis 3 | 94% 4 |
| Gene-targeted deletion/duplication analysis 5 | 2%-3% 6, 7 |
See Table A. Genes and Databases for 染色体位点 and protein.
2.
See Molecular Genetics for information on allelic variants detected in this 基因.
3.
Sequence analysis detects variants that are benign, likely benign, of 意义不确定, likely pathogenic, or pathogenic. Pathogenic variants may include small intragenic deletions/insertions and 错义, nonsense, and 剪接位点 variants; typically, 外显子 or whole-基因 deletions/duplications are not detected. For issues to consider in interpretation of 序列分析 results, click here.
4.
Caciotti et al [2015]. Intronic alterations account for 9% of 基因 alterations, the majority of which affect one of the -1,-2,+1, or +2 nucleotides [Morrone et al 2014].
5.
Gene-targeted deletion/duplication analysis detects intragenic deletions or duplications. Methods that may be used can include: quantitative PCR, long-range PCR, multiplex ligation-dependent probe amplification (MLPA), and a 基因-targeted microarray designed to detect single-外显子 deletions or duplications.
6.
Approximately 2.7% of pathogenic variants are secondary to large deletions [Caciotti et al 2015].
7.
One individual had maternal uniparental isodisomy of the telomeric end of 染色体 16, leading to disease [Catarzi et al 2012] and two individuals had large deletions including GALNS exons 10-14 and exons 9-14 [Caciotti et al 2015]; this finding may be detected by certain 染色体芯片 (CMA) methods.
N-acetylgalactosamine 6-sulfatase (GALNS) enzyme activity can be used when:
- Clinical findings strongly indicate MPS IVA and urine GAG analysis is normal;
AND/OR - Molecular genetic testing fails to identify 双等位基因的 pathogenic variants in GALNS.
In addition, establishing a diagnosis of MPS IVA by enzyme analysis may aid in interpretation of sequencing variants of 意义未明. GALNS enzyme activity can be measured in cultured fibroblasts or leukocytes. Because each laboratory has its own normal range of enzyme activity, results from different laboratories cannot be directly compared. The level of residual enzyme activity may correlate with disease severity.
Note:
1.Low enzyme activity can be caused by other disorders including:
- Multiple sulfatase deficiency (MSD), i.e., deficient activity of several sulfatases (including GALNS). Thus, when GALNS enzyme activity is abnormal, additional sulfatase enzymes need to be assayed to evaluate for MSD.
- Mucolipidosis II and mucolipidosis III (see ML III Alpha/Beta, ML III Gamma). Mucolipidosis II and mucolipidosis III impair mannose-6-phosphate lysosomal enzyme targeting, leading to low lysosomal enzyme activities in fibroblasts, high lysosomal enzyme activities in plasma, and relatively unchanged lysosomal enzyme activities in leukocytes.
2.
Because the clinical manifestations of MPS IVA and MPS IVB are indistinguishable, it is customary to measure B-galactosidase enzyme activity at the same time.
Clinical Characteristics
Clinical Description
Mucopolysaccharidosis type IVA (MPS IVA) comprises a clinical continuum ranging from a severe and rapidly progressive form to a slowly progressive form. In the past, the two forms were distinguished by height, the subjective assessment of severity of bone deformity, and survival [Tomatsu et al 2011]; however, neither clinical nor biochemical findings can provide clear distinctions between the two forms and, thus, the MPS IVA 表型 should be considered a continuum from severe to slowly progressive.
Affected children have no distinctive clinical findings at birth. The severe form is usually apparent between ages one and three years. The slowly progressive form may not become evident until late childhood or adolescence [Montaño et al 2007].
In both forms, the initial presentations vary and may be a single finding or several findings. Kyphoscoliosis, knock-knee (genu valgum) (Figure 5), and pectus carinatum (Figure 3 and Figure 4) are the most common initial manifestations of the severe form [Montaño et al 2007]. In contrast, hip problems including pain and stiffness (due to collapse and flattening of the proximal femoral epiphysis) are common initial manifestations of the slowly progressive form [Hecht et al 1984, Wraith 1995, White et al 2014].
Because descriptions of the natural history of MPS IV published in the past may not have distinguished between MPS IVA (Morquio syndrome type A; accounting for >95% of 受累的 individuals) and MPS IVB (Morquio syndrome type B; <5% of affected individuals), the following information is relevant to both MPS IVA and MPS IVB.
While the skeletal findings of MPS IVA are the hallmark findings, involvement of other organ systems can lead to significant morbidity, including respiratory compromise, obstructive sleep apnea, valvular heart disease, hearing impairment, corneal clouding, dental abnormalities, and hepatomegaly. Compression of the spinal cord results in neurologic involvement especially when disease is recognized later in life [reviewed in Neufeld & Muenzer 2001, Tomatsu et al 2011, Solanki et al 2013].
Coarse facial features are also present but milder than in other mucopolysaccharidoses (see Differential Diagnosis).
Children with MPS IVA typically have normal intellectual ability.
Ligamentous laxity and joint hypermobility are distinctive features of MPS IVA, and are rare among other storage disorders.
Musculoskeletal
Skeletal findings worsen over time. The combination of bone and joint involvement leads to pain and arthritis that result in subsequent disability.
Upper extremity involvement is also progressive and can impair hand-wrist strength and limit ability to perform some activities of daily living, such as using a fork. Hypermobility and ulnar deviation of the wrist joint are distinctive features of MPS IVA.
Lower extremity involvement, which is universal and progressive if untreated [Holzgreve et al 1981, Dhawale et al 2012], generally consists of malalignment due to progressive hip subluxation and/or valgus deformity. It can lead to significant gait alteration; hip, knee, and/or ankle pain with activity; and decreased endurance [Dhawale et al 2012].
Knee and ankle valgus are the most common lower extremity deformities. Genu valgum (knock-knee) results from distal femoral and proximal tibial involvement and joint laxity.
A longitudinal study using the Pediatric Evaluation of Disability Inventory and the Functional Independence Measure found severely limited joint mobility in persons with MPS IVA, generally with loss of ambulation late in the disease course. Aggressive and long-term intervention by a team of physical therapists and rehabilitative specialists is often needed to optimize mobility (see Management) [Guarany et al 2012].
Hip dysplasia. Early in the disease course the capital femoral epiphyses are small and the acetabuli are shallow. Subsequent progressive destructive changes in the femoral head and acetabuli result in hip dislocation, arthritis, and severe joint restriction, causing 受累的 individuals to become wheel-chair bound [reviewed in Tomatsu et al 2011].
Spinal cord compression may occur in any spinal segment; cervical spinal compression is the most common site. Spinal cord compression can be caused by cervical instability, unossified fibrocartilage associated with an abnormal odontoid process, ligamentous laxity, cartilaginous and ligamentous hypertrophy at the atlantoaxial joint, glycosaminoglycan (GAG) deposition in the extradural space, disc protrusion, thoracolumbar kyphosis, and acquired central canal stenosis [Lipson 1977, Ransford et al 1996, Tomatsu et al 2011, McKay et al 2012, Solanki et al 2013].
Odontoid hypoplasia leading to atlantoaxial instability, which later may result in upper cervical spinal cord compression, occurs in 90% of 受累的 individuals (Figure 6) [Hughes et al 1997].
Persons with untreated atlantoaxial instability often do not survive beyond the second or third decade because minor falls and/or neck extension can result in quadriparesis or sudden death [reviewed in Tomatsu et al 2011].
Spinal canal stenosis may be diffuse or focal. The causes of spinal canal stenosis are similar to the causes of spinal cord compression and include: kyphosis, disc protrusion, generalized thickening of posterior longitudinal ligament, or localized thickening of intervertebral ligaments due to GAG deposition.
Lumbar spine misalignment (i.e., thoracolumbar kyphosis) in older individuals can result in focal spinal stenosis, compressive myelopathy, and paraplegia [Dalvie et al 2001].
Ligamentous laxity resulting in hypermobile joints is common; however, decreased joint mobility can be observed in the large joints including knees, hips, and elbows [Neufeld & Muenzer 2001].
Neurologic
At the time of diagnosis, individuals with MPS IV (i.e., MPS IVA and MPS IVB) typically have normal developmental milestones and normal intellectual abilities. The neurologic findings of MPS IV are most often secondary to spinal abnormalities in the neck and/or lumbar region. The increased risk for neurologic compromise makes developmental delay and learning disabilities more common in children with MPS IVA than in unaffected children [Montaño et al 2007].
Subtle abnormal brain MRI findings such as prominent perivascular space, enlarged lateral ventricles, and prominent frontal CSF were reported in eight of 14 individuals with MPS IVA [Davison et al 2013]. From the same study, neurocognitive evaluation revealed behavioral issues including anxiety, depression, decreased attention span, and somatic complaints. Whether these findings are caused by disease-specific biochemical abnormalities, chronic illness, or a combination of the two is unknown.
Cardiac
Cardiac complications include ventricular hypertrophy and early-onset, severe valvular involvement. Coronary intimal sclerosis has also been reported [reviewed by Hendriksz et al 2013].
In a multicenter, multinational, cross-sectional study (MorCAP) involving 325 individuals with MPS IVA, valvular regurgitation was more common than valvular stenosis. Among those with valvular regurgitation, tricuspid regurgitation was the most common (35%). Mitral regurgitation, aortic regurgitation, and pulmonary regurgitation were found in 25%, 19%, and 14% of 受累的 individuals, respectively [Harmatz et al 2013]. Individuals with MPS IVA usually have an abnormally high heart rate and high myocardial index to compensate for small left ventricular diameter and low stroke volume [Hendriksz et al 2015].
Respiratory
Respiratory complications are a major cause of morbidity/mortality. Airway obstruction, sleep-disordered breathing, and restrictive lung disease have been described.
Glycosaminoglycan (GAG) accumulation in the adenoids, tonsils, pharynx, larynx, trachea, and bronchial tree leads to adenotonsillar hypertrophy, tracheal distortion, trachea- and bronchomalacia, and obstructive sleep apnea [Semenza & Pyeritz 1988, Walker et al 2003]. Deposition of GAG in the trachea and bronchi could cause tortuosity of the airway leading to buckling and airway obstruction when the neck is flexed. If not recognized (particularly during cervical spine fusion/stabilization surgery) and if the head and neck are fused in a flexed position, acute obstruction may result on tracheal extubation [reviewed in Solanki et al 2013].
Restrictive lung disease results from a small thorax, chest wall anomalies, spine deformities, neuromuscular compromise from cervical myelopathy, and hepatomegaly causing upward displacement of the diaphragm [Walker et al 2003, Hendriksz et al 2013].
Because of atlantoaxial instability and upper-airway obstruction, persons with MPS IV prefer to sleep prone on a flat surface without a pillow in order to keep the neck extended and minimize the tortuosity of the airway.
If respiratory complications are not recognized or are not treated, cor pulmonale and respiratory failure can ensue, leading to early death [Semenza & Pyeritz 1988, Walker et al 2003, Montaño et al 2007, Hendriksz et al 2013].
Growth
Children with MPS IVA have a normal birth weight and a longer than normal birth length.
Between ages one and three years the growth velocity decreases compared to unaffected children.
By age 18 years, the average height in males is 123 cm and in females 117 cm, compared to 177 cm and 163 cm in unaffected males and females, respectively.
Eye
Ophthalmologic findings are present in more than 50% of individuals with MPS IVA. Natural history studies have not been performed; thus, it is not possible to predict the age of onset of ophthalmologic findings [reviewed in Wood et al 2013].
Slowly progressive corneal clouding, found in 50% of 受累的 individuals (ages 1-65 years), is the most common ophthalmologic finding in MPS IVA.
Other less common ophthalmologic findings include: astigmatism, cataracts, punctate lens opacities, open-angle glaucoma, optic disc swelling, optic atrophy, and retinopathy. While rare, these ophthalmologic findings can be serious secondary complications [reviewed in Hendriksz et al 2013, Hendriksz et al 2015].
Pseudoexophthalmos, the appearance of a bulging eye secondary to a shallow orbit, can cause exposure keratitis and also be of cosmetic concern.
Dental
Deciduous teeth erupt normally and are widely spaced and discolored with thin irregular (stippled) enamel and small pointed cusps which flatten over time with normal wear.
Permanent teeth also have hypoplastic enamel [reviewed in Onçağ et al 2006].
Hearing
Mild to moderate hearing loss is common in MPS IVA. Hearing impairment is often noted toward the end of the first decade.
Mixed (i.e., combined conductive and sensorineural) hearing loss is more common than conductive or sensorineural hearing loss alone.
Conductive hearing loss is secondary to recurrent middle ear infections, serous otitis media, and deformity of the ossicles [Hendriksz et al 2013].
Sensorineural hearing loss secondary to GAG accumulation in the inner ear and/or central nervous system has been described [Ruckenstein et al 1991, Simmons et al 2005, Hendriksz et al 2013].
Genotype-Phenotype Correlations
Genetic alterations in GALNS that are predicted to severely affect protein function, such as deletions and nonsense variants, are common in individuals with rapidly progressive growth failure [Montaño et al 2007, Morrone et al 2014].
In contrast, 受累的 individuals with more conservative genetic changes, such as p.Thr312Ser, are more likely to have a milder 表型, likely due to retained partial enzymatic activity [Yamada et al 1998].
Individuals 纯合性 for c.898+1G>C generally have a slowly progressive course [Tomatsu et al 2005].
Nomenclature
Much of the older literature and more complete natural history studies were performed prior to understanding the basis of MPS IVA (N-acetylgalactosamine 6-sulfatase deficiency or 双等位基因的 GALNS pathogenic variants) and MPS IVB (B-galactosidase deficiency or biallelic GLB1 pathogenic variants). Thus, the term MPS IV refers to individuals with a clinical diagnosis without an enzymatic and/or molecular diagnosis, whereas the more specific terms MPS IVA and MPS IVB refer to individuals with an enzymatic and/or molecular diagnosis.
Mucopolysaccharidosis type IVA (MPS IVA), also known as Morquio syndrome type A, was initially characterized by Morquio [1929] and Brailsford [1929].
MPS IVA and MPS IVB are known as Morquio syndrome type A and type B, respectively.
Prevalence
MPS IVA is rare. The prevalence in Australia has been estimated at 1:926,000, whereas the prevalence in the UK has been estimated at 1:599,000. The birth prevalence for MPS IVA ranged from 1:71,000 to 1:179,000 across multiple countries [Leadley et al 2014]. In one German study, the incidence of MPS IVA was 1:270,000 and that of MPS IVB less than 1:1,000,000 [Baehner et al 2005]. Similarly, the incidence of MPS IVA in Italy was estimated at 1:300,000 live births [Caciotti et al 2015].
Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview are associated with pathogenic variants in GALNS.
Differential Diagnosis
Mucopolysaccharidosis type IVB(MPS IVB). The disorder that most closely resembles mucopolysaccharidosis type IVA (MPS IVA) is MPS IVB, in which accumulation of keratan sulfate occurs due to 双等位基因的 pathogenic variants in GLB1, the 基因 encoding the enzyme B-galactosidase. In most individuals with clinical findings of MPS IV, MPS IVA can be distinguished from MPS IVB only by biochemical testing and/or 分子遗传学检测.
Biallelic pathogenic variants in GLB1 also cause GM1 gangliosidosis, a lysosomal storage disease with severe neurologic outcomes and skeletal dysplasia. The spectrum of GM1 gangliosidosis comprises a continuum of infantile, late infantile, juvenile, and adult forms. The infantile form often results in death by age two years, while life span may not be shortened in the adult form.
Although novel GLB1 pathogenic variants identified in MPS IVB often map to the substrate binding regions of the protein [reviewed in Ohto et al 2012], some pathogenic variants are associated with both GM1 gangliosidosis and MPS IVB.
Other mucopolysaccharidoses. Signs and symptoms of MPS IVA overlap with those of other mucopolysaccharidoses, all of which have a broad spectrum of clinical manifestations. See MPS I and MPS II.
Compared to other mucopolysaccharidoses, MPS IVA is characterized by normal intellectual abilities and less coarsening of the facial features. In general, visual acuity in persons with MPS IVA is better than that of persons with other types of MPS. In addition, joint hypermobility is unique to MPS IV. Atlanto-axial instability is more common in individuals with MPS IV than in those with the other mucopolysaccharidoses.
See Mucopolysaccharidoses: OMIM Phenotypic Series to view genes associated with this 表型 in OMIM.
Spondyloepiphyseal dysplasia (SED) has similar radiographic findings. Clinical features, especially extraskeletal features, could distinguish SED from MPS IVA. See Schimke Immunoosseous Dysplasia and X-Linked Spondyloepiphyseal Dysplasia Tarda.
Legg-Calve-Perthes disease (OMIM). In some instances mild MPS IVA can manifest only with hip pain at the onset of disease, which can lead to an initial misdiagnosis of Legg-Calve-Perthes disease.
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with mucopolysaccharidosis type IVA (MPS IVA), the following evaluations are recommended [Hendriksz et al 2013, Solanki et al 2013, Hendriksz et al 2015]:
- Baseline neurologic examination to assess for signs of spinal cord compression
- Plain radiographs
- Baseline radiographs of the cervical spine, including anterior-posterior (AP), neutral lateral, and flexion-extension views
- Baseline AP and lateral radiographs of the entire spine
- Baseline AP and frog leg lateral radiographs of the pelvis
- Baseline AP standing radiographs of the lower extremities; in individuals who are able to stand independently and have signs of lower extremity misalignment, baseline standing lower extremity alignment radiograph from hip to ankle
- MRI
- Baseline MRI of the entire spine (neutral position) focusing on potential sites of cord compression (occipitocervical, cervicothoracic, and thoracolumbar)
- Flexion-extension MRI of the cervical spine when clinically indicated – for example, if cervical spine instability is suspected on plain radiographs:
- Because MRI is usually performed under sedation or anesthesia, cervical spine radiographs should be obtained prior to anesthesia to evaluate for upper cervical spine instability.
- In individuals with cervical spine instability suspected on plain film or those with inconclusive radiographic findings, flexion-extension cervical spine MRI can be used to evaluate for cord compression.
- Flexion-extension MRI can identify dynamic changes in canal diameter leading to cord compression.
- Mackenzie et al [2013] demonstrated that cervical spine flexion-extension MRI under sedation/anesthesia in children with skeletal dysplasia was safe under adequate supervision and the result was useful for surgical decision making.
- Evaluation by:
- A physiatrist (i.e., specialist in physical medicine and rehabilitation [PM&R]) to assess mobility and autonomy. Endurance tests including six-minute walk test (6MWT) or timed 25-foot walk test (T25FW) in patients with limited ambulation have been used to evaluate functional status of the cardiovascular, pulmonary, musculoskeletal, and nervous systems. 6MWT and T25FW should be performed at the time of diagnosis.
- A physical therapist to assess joint range of motion and mobility
- An occupational therapist to assess activities of daily living such as the functional dexterity test (FDT). Baseline upper extremity strength assessments should be performed, including grip strength assessed by dynamometer and pinch strength assessed by a pinch meter [White et al 2014].
- Consultation with a clinical geneticist and/or genetic counselor
- Electrocardiogram and echocardiogram. If coronary artery involvement is suspected, noninvasive stress imaging should be performed [Braunlin et al 2011, Hendriksz et al 2013].
- Baseline pulmonary function and polysomnography, with evaluation by a pulmonologist
- Audiology evaluation
- Dental evaluation
- Ophthalmology evaluation to assess visual acuity and intraocular pressure; slit lamp examination and posterior segment examination for retinopathy
- Plotting of height and weight on growth charts specific for MPS IVA [Montaño et al 2007, Montaño et al 2008]
- Pain assessment and quality of life questionnaires
Treatment of Manifestations
Management of individuals with MPS IVA is best undertaken by the following multiple specialists, coordinated by a physician specializing in the care of individuals with complex medical problems:
- Physiatrist (specialist in physical medicine and rehabilitation [PM&R]) to optimize mobility and autonomy
- Physical therapist to optimize mobility
- Occupational therapist to optimize autonomy
- Psychological support to optimize coping skills and quality of life
- Educational professions to optimize learning in a medically fragile individual
- Consideration of referral to family therapy to help normalize the experience for the 受累的 individual, parents, sibs, and extended family members
- Home care for 受累的 individuals with multiple medical equipment needs
- Hospice for end-of-life care
Enzyme Replacement Therapy (ERT)
Recombinant human GALNS ERT (elosulfase alfa, or Vimizim™) was approved by the FDA in February 2014.
- The recommendation dose is 2 mg/kg/week intravenous. Although the treatment with ERT is not curative, ERT could improve endurance and overall quality of life.
- Premedication (30-60 minutes prior to each enzyme infusion) with a non-sedating antihistamine (if possible) with or without antipyretics is recommended to prevent infusion-associated reactions.
- A Phase 3 clinical trial demonstrated a statistically significant improvement in the 6MWT distance in the 2 mg/kg weekly dose group compared to the placebo group. The 3-minute stair climb test (3MSCT) and respiratory function were improved with the treatment but the differences were not statistically significant.
- The long-term effects of this treatment on the skeletal features of MPSIVA are not yet known (see Therapies Under Investigation). The efficacy of ERT in improving pathology in the musculoskeletal system may be limited because of poor biodistribution of the enzyme in avascular tissue.
Musculoskeletal
For published orthopedic management guidelines, see White et al [2014]. The level of physical activity should be monitored by specialists in orthopedic surgery, neurology, physiatry, and physical therapy to optimize mobility while preventing joint injury, joint misalignments, and cervical cord damage.
Upper extremities. Non-operative interventions, such as external wrist splints, may be considered. Surgical intervention, including partial or complete wrist fusion, may be necessary to stabilize wrist range of motion.
Knee and ankle valgus. Lower extremity malalignment associated with progressively poor mechanical alignment and decreasing endurance requires intervention; however, no absolute indications for intervention exist.
- Growth modulation, also called guided growth (temporary surgical tethering of the growth plate to allow gradual correction of the deformity) or realignment osteotomies have been successful [Dhawale et al 2012, White et al 2014].
- Early detection and evaluation may allow surgical tethering of the growth plate to treat mild-to-moderate lower extremity angular deformities in children with open physes (growth plates). Typically this procedure is less invasive and allows for easier recovery than realignment osteotomies.
- Once the growth plates close, distal femoral and proximal tibial osteotomies are needed to acutely or gradually (with the use of external fixators) correct lower extremity malalignment.
- Ankle malalignment is often corrected by a distal tibial osteotomy with distal tibial screw hemiepiphyseodesis [reviewed in Tomatsu et al 2011].
Hip dysplasia. Surgery can manage pain and alignment and permit optimal mobility.
- Hip reconstruction includes either femoral or acetabular osteotomy for mild cases or combined acetabular and femoral osteotomy for severe cases. Augmentation of acetabular bone stock and customized implants by using cortical grafts from the inner table of the ilium are usually required due to a shallow acetabulum [White et al 2014].
- Total hip arthroplasty may be required in young adults experiencing significant hip pain which cannot be corrected by reconstructive techniques.
Odontoid hypoplasia. When upper cervical spine instability is documented or when clinical findings of cervical myelopathy are present, occipito-cervical or upper cervical decompression and fusion are required to stabilize the upper cervical spine and relieve cervical cord compression.
- To minimize neurologic injury and maximize function, intervention in children is recommended when radiographic signs of cervical compression are present, even in the absence of symptoms.
- Affected individuals undergoing surgical fusion typically do well; minor secondary complications can include pin site infections, pressure sores, and long-term difficulty with endotracheal intubation.
- Note: It is important for clinicians to be aware that cervical myelopathy from upper cervical instability may result in deteriorating endurance and worsening gait. If myelopathy is suspected, obtain cervical spine radiographs and MRI (see Surveillance). The 受累的 individual should be referred for evaluation by a pediatric orthopedic surgeon or neurosurgeon at a tertiary care facility.
Lumbar spine malalignment. Thoracolumbar kyphosis (resulting from vertebral hypoplasia) may be progressive and symptomatic.
- When kyphosis is less than 45 degrees, the risk of progressive deformity is less than with a greater curve, but warrants clinical and radiographic monitoring.
- When kyphosis exceeds 45 degrees, progression is likely. Although extensive bracing with an orthosis or a cast does not prevent progression of the thoracolumbar kyphosis, it may delay the need for surgical intervention during a period of growth and development.
- Anterior and posterior circumferential spinal fusion are indicated if one or more of the following are present:
- Progressive thoracolumbar kyphosis greater than 70 degrees
- Uncontrolled back pain
- Neurologic changes related to spinal stenosis
Cardiac
Elevated heart rates could indicate a compensation mechanism, secondary to small left ventricular diameter and small stroke volume; thus tachycardia treatment with beta blockers should be avoided. Valve replacement may be considered for progressive valvular problems [Hendriksz et al 2015]. Risks need to be carefully weighed for valve replacement, either mechanical (life-long use of anticoagulants) or bioprosthesis (increased risk of valve dysplasia, degradation, and calcification).
Respiratory
Upper-airway obstruction and obstructive sleep apnea are managed by removal of enlarged tonsils and adenoids (average age 7 years [Montaño et al 2007]). Note: Even with this intervention, the rate of obstructive sleep apnea in children with a mucopolysaccharidosis is much higher than the general population; therefore, prompt clinical evaluation and referral for polysomnography are appropriate [Nashed et al 2009].
In persons with diffuse narrowing of the airway in whom adenotonsillectomy only temporally relieves upper-airway obstruction, other interventions to consider are: CPAP (continuous positive airway pressure), BiPAP (bilevel positive airway pressure), and tracheostomy.
Lower-airway obstruction manifest as wheezing and recurrent infection is managed by inhaled and/or oral bronchodilators and, in some instances, corticosteroids.
Restrictive lung disease is managed by supportive treatment.
Growth
Height of children with MPS IVA is best plotted on growth charts specific for MPS IVA [Montaño et al 2007, Montaño et al 2008].
Nutrition should be optimized with a balanced diet and adequate vitamin D and calcium to assure bone health.
Learning Environment
Despite some physical limitation, individuals with MPS IVA have normal intellect and can thrive in an environment with academic and social stimulation. Children routinely attend regular class/school with assistance to prevent physical injury.
Eye
Corneal opacification often causes reduced vision in early childhood, necessitating penetrating keratoplasty, for which the outcome can vary. Recurrence of opacities within the first year post keratoplasty has been reported, making this a temporary measure for improving quality of life [reviewed by Bothun et al 2011]. In addition, other ophthalmologic problems including glaucoma and retinopathy may limit the success of corneal transplantation. Cataract surgery may benefit those with cataracts.
Dental
Daily oral hygiene care, fissure sealing, and adequate fluoride supplementation help prevent cavities. Orthodontic management to correct malocclusion may be necessary.
Hearing
Because ventilation tube placement can minimize the long-term scarring associated with chronic middle-ear effusions and recurrent acute otitis media, and improve hearing in the long term, most children have ventilation tubes placed during the preschool years. At the first occasion, a long-lasting tympanostomy tube is recommended due to high risk of recurrent middle-ear effusion and the risk associated with sedation in individuals with MPS IVA [Hendriksz et al 2015].
The progressive hearing impairment observed in most individuals with MPS IVA benefits from hearing aids.
Prevention of Primary Manifestations
See Treatment of Manifestations for information regarding ERT.
The experience of hematopoietic stem cell therapy is very limited and has not been well studied.
Prevention of Secondary Complications
Anesthesia. Procedures requiring anesthesia require considerable planning and are best performed in a facility in which anesthesiologists are experienced with the airway issues of MPS IVA, such as abnormal anatomy and GAG accumulation, unstable cervical spine, and progressive pulmonary disease (both restrictive and obstructive). Theroux et al [2012], who published the largest cohort of children with Morquio syndrome undergoing anesthesia, made specific recommendations for care during anesthesia.
Preoperative assessment should include history of response to anesthesia and any evidence of airway obstruction; cardiac evaluation, including electrocardiogram and echocardiography; evaluation of respiratory function (spirometry and polysomnography); and airway fluoroscopy [Muhlebach et al 2011, Tomatsu et al 2011].
Endotracheal intubation likely includes use of a video laryngoscope, fiberoptic bronchoscope with or without a laryngeal mask airway, and a smaller endotracheal tube than expected for age or size. Although nasal intubation is an option, GAG deposits can lead to narrowing of the nasal passages and increased propensity to bleeding [Aziz et al 2011, Muhlebach et al 2011, Walker et al 2013].
Postoperative narcotic management should be judicious; multimodal analgesics and non-narcotic medications are preferable to avoid exacerbating preexisting respiratory issues, such as sleep apnea.
Postoperative complications including pulmonary edema have been described [Morgan et al 2002].
Surgery. Because subacute spinal stenosis and/or dynamic spinal stenosis could lead to spinal cord injury, procedures involving cervical spine manipulation, prone positioning (including spinal surgery), and/or prolonged time under anesthesia (e.g., exceeding 45 minutes), should be considered for intraoperative neurophysiologic monitoring (IONM). IONM uses somatosensory and motor evoked potentials (SSEPs or MEPs) to monitor spinal cord function [Solanki et al 2013, Walker et al 2013]. Note: While spinal infarct during surgery was reported in a few individuals with skeletal dysplasia [Tong et al 2012, Pruszczynski et al 2015], data demonstrating consistent improvement of outcome with this monitoring technology are limited [Solanki et al 2013].
Immunizations. Due to increased risk for pulmonary infection, all 受累的 individuals should receive influenza and pneumococcal immunizations as well as routine immunizations.
Bacterial endocarditis prophylaxis is recommended for those at high risk, including those with a prosthetic cardiac valve, prosthetic material used for cardiac valve repair, or previous infective endocarditis [Wilson et al 2007].
Surveillance
Individuals on ERT
The following should be assessed before and after initiation of ERT to determine treatment efficacy:
- Physical and neurologic evaluation at least every six months
- Annual assessment of quality of life, disease burden, and endurance (see All Individuals)
- Annual pulmonary function tests, including maximum voluntary ventilation (MVV) and forced vital capacity (FVC)
Note: (1) Changes in urine keratan sulfate (KS) levels do not correlate with efficacy of treatment; therefore, the benefit of following urine KS (or urine GAG) levels during elosulfase alfa treatment is limited. (2) The benefit of monitoring anti-elosulfase alfa antibodies is unknown.
All Individuals
Assessment of quality of life, disease burden, and endurance
- Every 6-12 months: track growth, pubertal stage, and progress; optimize ambulation
- Pain severity assessment every six months; age-appropriate quality of life questionnaires every year
- Yearly, before and after surgical procedures, or as clinically indicated: endurance tests including six-minute walk test (6MWT) or timed 25-foot walk (T25FW) to evaluate functional status of the cardiovascular, pulmonary, musculoskeletal, and nervous systems. Respiratory rate, pulse oximeter, and heart rate should be measured before and after the annual testing.
Musculoskeletal.White et al [2014] recommended the following guidelines for monitoring musculoskeletal involvement in those with MPS IVA (full text):
- Upper and lower extremities. Evaluate severity and progression of upper and lower extremity involvement at least annually.
- Evaluation of range of motion, grip and pinch strength, and functional assessments (e.g., functional dexterity test) of the upper extremities
- Assessment of lower extremity alignment, including standing AP radiographs (as clinically indicated) and AP and frog leg lateral radiographs of the pelvis to assess hip dysplasia/subluxation until skeletal maturity or when clinically indicated
- Spine. Neurologic examination every six months to assess for spinal cord compression [Solanki et al 2013]:
- For children who are reliable historians, at each clinic visit obtain a history of exercise tolerance and symptoms of myelopathy (e.g., extremity weakness; clumsiness; unsteady, changing gait; bowel or bladder dysfunction or lower back/leg pain).
- In those with multisegmental myelopathy, SSEPs and MEPs (if available) may provide detailed information.
- Perform plain radiography of the cervical spine (AP, lateral, neutral, and flexion-extension) every six months.
- Perform plain radiography of the spine (AP and lateral views for the thoracolumbar spine) every one to three years.
- Perform MRI whole spine (neutral position)* annually and flexion-extension MRI of the cervical spine every one to three years.
* Neutral, flexion, and extension lateral radiographs of the cervical spine should be obtained prior to cervical MRI to assess for atlanto-occipital instability.
Cardiac. Evaluate heart rate annually; perform electrocardiogram and echocardiogram every one to three years depending on disease course [Braunlin et al 2011, Hendriksz et al 2013, Hendriksz et al 2015].
Respiratory
- For obstructive sleep apnea, yearly history focused on sleep patterns and sounds. Evaluation by an otolaryngologist for adenotonsillectomy. Annual in home screening sleep studies (which monitor oxygen saturation). Polysomnography every three years.
- To assess pulmonary function, annual MVV and FVC until children stop growing, then every two to three years. The benefit of noninvasive pulmonary function tests, impulse oscillometry, and thoracoabdominal motion analysis has been demonstrated in children with MPS IV [Rodriguez et al 2010].
Growth. Use MPS IVA-specific growth charts to monitor nutritional status [Montaño et al 2007, Montaño et al 2008]. Length/height and weight should be measured at every visit.
Eye
- Monitor for vision and ocular abnormalities at every visit; refer to an ophthalmologist as needed.
- For those with rod and cone retinal dystrophy, perform retinal examination and electroretinography (ERG) under scotopic and photopic conditions every five years [Hendriksz et al 2013].
Dental. Evaluate every six to 12 months.
Hearing. Perform yearly audiogram.
Agents/Circumstances to Avoid
Because excessive weight gain causes undue stress on the axial skeleton and may decrease the duration of independent ambulation, it is important to optimize nutrition for growth while maintaining a lean habitus.
Due to small ventricular diameter and stroke volume, beta blockers should be avoided in the treatment of tachycardia.
Evaluation of Relatives at Risk
It is appropriate to evaluate apparently asymptomatic younger sibs of a 先证者 in order to identify as early as possible those who would benefit from initiation of ERT (see Treatment of Manifestations). Evaluations can include:
- Molecular genetic testing if the pathogenic variants in the family are known;
- Analysis of N-acetylgalactosamine 6-sulfatase (GALNS) enzyme activity if the pathogenic variants in the family are not known.
See Genetic Counseling for issues related to testing of at-risk relatives for 遗传咨询 purposes.
Therapies Under Investigation
ERT (elosulfase alfa, or Vimizim™) use in mildly 受累的 individuals is still being investigated. As reported in the MOR-008 Phase 2 clinical trial in which all participants were high functioning (defined as a mean baseline walking distance of 70%-80% of the unaffected population), ERT made little change in endurance and respiratory function tests but some positive change in exercise capacity, muscle strength, and pain. Hypersensitivity reactions occurred in 18.7% of participants. Over all, the safety profile of elosulfase alfa appears manageable and serious adverse reactions are uncommon, but further long-term safety data are being collected [Tanpaiboon 2015].
Search ClinicalTrials.gov for access to information on clinical studies for a wide range of diseases and conditions.
Genetic Counseling
Genetic counseling is the process ofproviding individuals and families with information on the nature, inheritance,and implications of genetic disorders to help them make informed medical andpersonal decisions. The following section deals with genetic risk assessment andthe use of family history and genetic testing to clarify genetic status forfamily members. This section is not meant to address all personal, cultural, orethical issues that individuals may face or to substitute for consultation witha genetics professional. —ED.
Mode of Inheritance
Mucopolysaccharidosis type IVA (MPS IVA) is inherited in an 常染色体隐性遗传 manner.
Risk to Family Members
Parents of a 先证者
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Sibs of a 先证者
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Offspring of a 先证者. The offspring of an individual with MPS IVA are obligate heterozygotes (carriers) for a GALNS 致病性变异.
Other family members. Each sib of the 先证者’s parents is at a 50% risk of being a 携带者 of a GALNS 致病性变异.
Carrier (Heterozygote) Detection
Carrier testing for at-risk relatives requires prior identification of the GALNS pathogenic variants in the family.
Related Genetic Counseling Issues
See Management, Evaluation of Relatives at Risk for information on evaluating at-risk relatives for the purpose of early diagnosis and treatment.
Family planning
- The optimal time for determination of genetic risk, clarification of 携带者 status, and discussion of the availability of prenatal testing is before pregnancy.
DNA banking is the storage of DNA (typically extracted from white blood cells) for possible future use. Because it is likely that testing methodology and our understanding of genes, allelic variants, and diseases will improve in the future, consideration should be given to banking DNA of 受累的 individuals.
Prenatal Testing and Preimplantation Genetic Diagnosis
Once the GALNS pathogenic variants have been identified in an 受累的 family member, prenatal testing and 植入前遗传诊断 for a pregnancy at increased risk for MPS IVA are possible options.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing, particularly if the testing is being considered for the purpose of pregnancy termination rather than early diagnosis. Although decisions about prenatal testing are the choice of the parents, discussion of these issues is appropriate.
Resources
GeneReviews staff has selected the following disease-specific and/orumbrella support organizations and/or registries for the benefit of individualswith this disorder and their families. GeneReviews is not responsible for theinformation provided by other organizations. For information on selectioncriteria, click here.
- Canadian Society for Mucopolysaccharide and Related Diseases, Inc.PO Box 30034North Vancouver British Columbia V7H 2Y8CanadaPhone: 800-667-1846 (toll free); 604-924-5130Fax: 604-924-5131Email: info@mpssociety.ca
- Medline Plus
- Morquiosity
- My46 Trait Profile
- National Library of Medicine Genetics Home Reference
- National MPS SocietyPO Box 14686Durham NC 27709-4686Phone: 877-677-1001 (toll-free); 919-806-0101Fax: 919-806-2055Email: info@mpssociety.org
- Society for Mucopolysaccharide Diseases (MPS)MPS House Repton PlaceWhite Lion RoadAmersham Buckinghamshire HP7 9LPUnited KingdomPhone: 0345 389 9901Email: mps@mpssociety.co.uk
- The Carol Ann Foundation & The International Morquio Organization8164 West Circulo De Los MorterosPO Box 64184Tucson AZ 85743Phone: 520-744-2531Email: mbs85705@yahoo.com; tomatsus@slu.edu
- National Organization for Rare Disorders (NORD)RareCareSMPhone: 800-999-6673
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. 鈥�ED.
Table A.
Mucopolysaccharidosis Type IVA : Genes and Databases
| Gene | Chromosome Locus | Protein | Locus-Specific Databases | HGMD | ClinVar |
|---|---|---|---|---|---|
| GALNS | 16q24 | N-acetylgalactosamine-6-sulfatase | GALNS database | GALNS | GALNS |
Table B.
OMIM Entries for Mucopolysaccharidosis Type IVA (View All in OMIM)
Pathophysiology
Mucopolysaccharidosis type IVA (MPS IVA) is caused by a deficiency of the lysosomal enzyme N-acetylgalactosamine-6- sulfatase (GALNS), which cleaves the keratan sulfate at the O-linked sulfate moiety of keratan sulfate (KS) and chondroitin-6-sulfate (C6S). The absence of the enzyme GALNS leads to intracellular accumulation of the glycosaminoglycans KS and C6S in the lysosomes of multiple tissues. The accumulation mainly in cornea and bone leads to the pathognomonic findings of corneal clouding and skeletal dysplasia [reviewed in Neufeld & Muenzer 2001].
GALNS
Gene structure.GALNS comprises 14 exons. For a detailed summary of 基因 and protein information, see Table A, Gene.
Pathogenic allelic variants. Missense, nonsense, and 剪接 variants, as well as small deletions, small insertions, gross insertions/duplications, and gross deletions have been found in GALNS.
Table 2.
GALNS Pathogenic Variants Discussed in This GeneReview
| DNA Nucleotide Change | Protein Amino Acid Change | Reference Sequences 1 |
|---|---|---|
| c.337A>T 2 | p.Ile113Phe | NM_000512 NP_000503 |
| c.898+1G>C | -- | |
| c.935C>G 3 | p.Thr312Ser |
Note on variant classification: Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
Note on nomenclature: GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen
鈥�.hgvs.org). See Quick Reference for an explanation of nomenclature. - 1.
Reviewed in Neufeld & Muenzer [2001]
- 2.
British-Irish variant
- 3.
British-Irish variant, mild 表型
Normal 基因产物. The enzyme N-acetylgalactosamine-6-sulfatase (GALNS) removes the terminal sulfate groups in mucopolysaccharides (e.g., KS and C6S). This 522-amino acid protein has a signal peptide of amino acids 1 through 26. The protein has at least one bound calcium in the catalytic 结构域. Processing of cysteine 79 to 3-oxoalanine by formylglycine-generating enzyme is targeted by the specific sequence CXPXR. This 3-oxoalanine is part of the catalytic site of all sulfatases.
Based on the finding that 26% of pathogenic 错义 variants were caused by transitional mutations at CpG dinucleotides, the role of 甲基化 in regulation of GALNS was identified: methylation was extensive within exons 2 through 14 [Tomatsu et al 2005].
GALNS consists of an N-terminal 结构域 including the active site, a second domain with antiparallel B-strands, and a C-terminal domain region which loops back to form a region of the active site. The GALNS active site contains a calcium bound to the catalytic nucleophile. Each GALNS molecule contains two glycosylation sites at residues Asn204 and Asn 423. Within each monomer are three disulfide bonds and one unpaired cysteine, which can pair with a second subunit to form the functional dimeric GALNS [Rivera-Colón et al 2012].
Abnormal 基因产物. The abnormal gene product demonstrates decreased enzymatic activity, leading to accumulation of sulfated intermediates. A GALNS 致病性变异 can lead to misfolding (and subsequent early degradation), mislocalization, or alterations in the catalytic 结构域 either directly or through substrate binding sites [Rivera-Colón et al 2012].
References
Literature Cited
- Aziz MF, Healy D, Kheterpal S, Fu RF, Dillman D, Brambrink AM. Routine clinical practice effectiveness of the Glidescope in difficult airway management: an analysis of 2,004 Glidescope intubations, complications, and failures from two institutions. Anesthesiology. 2011 Jan;114(1):34鈥�41. [PubMed: 21150569]
- Baehner F, Schmiedeskamp C, Krummenauer F, Miebach E, Bajbouj M, Whybra C, Kohlschütter A, Kampmann C, Beck M. Cumulative incidence rates of the mucopolysaccharidoses in Germany. J Inherit Metab Dis. 2005;28:1011鈥�7. [PubMed: 16435194]
- Bothun ED, Decanini A, Summers CG, Orchard PJ, Tolar J. Outcome of penetrating keratoplasty for mucopolysaccharidoses. Arch Ophthalmol. 2011;129:138鈥�44. [PubMed: 21320956]
- Brailsford J. Chondro-osteo-dystrophy: Roentgenographic anc clinical features of child with dislocation of vertebrae. Am J Surg. 1929;7:404鈥�10. [PubMed: 770041]
- Braunlin EA, Harmatz PR, Scarpa M, Furlanetto B, Kampmann C, Loehr JP, Ponder KP, Roberts WC, Rosenfeld HM, Giugliani R. Cardiac disease in patients with mucopolysaccharidosis: presentation, diagnosis and management. J Inherit Metab Dis. 2011;34:1183鈥�97. [PMC free article: PMC3228957] [PubMed: 21744090]
- Caciotti A, Tonin R, Rigoldi M, Ferri L, Catarzi S, Cavicchi C, Procopio E, Donati MA, et al. Optimizing the molecular diagnosis of GALNS: Novel methods to define and characterize Morquio A syndrome-associated mutations. Hum Mutat. 2015;36:357鈥�368. [PubMed: 25545067]
- Catarzi S, Giunti L, Papadia F, Gabrielli O, Guerrini R, Donati MA, Genuardi M, Morrone A. Morquio A syndrome due to maternal uniparental isodisomy of the telomeric end of chromosome 16. Mol Genet Metab. 2012;105:438鈥�42. [PubMed: 22178352]
- Dalvie S, Skinner J, Vellodi A, Noorden MH. Mobile thoracolumbar gibbus in Morquio type A: the cause of paraparesis and its management. J Pediatr Orthop B. 2001;10:328鈥�30. [PubMed: 11727377]
- Davison JE, Kearney S, Horton J, Foster K, Peet AC, Hendriksz CJ. Intellectual and neurological functioning in Morquio syndrome (MPS IVa). J Inherit Metab Dis. 2013;36:323鈥�8. [PubMed: 22231379]
- Dhawale AA, Thacker MM, Belthur MV, Rogers K, Bober MB, Mackenzie WG. The lower extremity in Morquio syndrome. J Pediatr Orthop. 2012;32:534鈥�40. [PubMed: 22706472]
- Guarany NR, Schwartz IV, Guarany FC, Giugliani R. Functional capacity evaluation of patients with mucopolysaccharidosis. J Pediatr Rehabil Med. 2012;5:37鈥�46. [PubMed: 22543891]
- Harmatz P, Mengel KE, Giugliani R, Valayannopoulos V, Lin SP, Parini R, Guffon N, Burton BK, Hendriksz CJ, Mitchell J, Martins A, Jones S, Guelbert N, Vellodi A, Hollak C, Slasor P, Decker C. The Morquio A Clinical Assessment Program: baseline results illustrating progressive, multisystemic clinical impairments in Morquio A subjects. Mol Genet Metab. 2013;109:54鈥�61. [PubMed: 23452954]
- Hecht JT, Scott CI Jr, Smith TK, Williams JC. Mild manifestations of the Morquio syndrome. Am J Med Genet. 1984;18:369鈥�71. [PubMed: 6431819]
- Hendriksz CJ, Al-Jawad M, Berger KI, Hawley SM, Lawrence R, Mc Ardle C, Summers CG, Wright E, Braunlin E. Clinical overview and treatment options for non-skeletal manifestations of mucopolysaccharidosis type IVA. J Inherit Metab Dis. 2013;36:309鈥�22. [PMC free article: PMC3590399] [PubMed: 22358740]
- Hendriksz CJ, Berger KI, Giugliani R, Harmatz P, Kampmann C, Mackenzie WG, Raiman J, Villarreal MS, Savarirayan R. International guidelines for the management and treatment of Morquio A syndrome. Am J Med Genet A. 2015;167A:11鈥�25. [PMC free article: PMC4309407] [PubMed: 25346323]
- Holzgreve W, Grobe H, von Figura K, Kresse H, Beck H, Mattei JF. Morquio syndrome: clinical findings in 11 patients with MPS IVA and 2 patients with MPS IVB. Hum Genet. 1981;57:360鈥�5. [PubMed: 6793501]
- Hughes DG, Chadderton RD, Cowie RA, Wraith JE, Jenkins JP. MRI of the brain and craniocervical junction in Morquio's disease. Neuroradiology. 1997;39:381鈥�5. [PubMed: 9189888]
- Leadley RM, Lang S, Misso K, Bekkering T, Ross J, Akiyama T, Fietz M, Giugliani R, Hendriksz CJ, Hock NL, McGill J, Olaye A, Jain M, Kleijnen J. A systematic review of the prevalence of Morquio A syndrome: challenges for study reporting in rare diseases. Orphanet J Rare Dis. 2014;9:173. [PMC free article: PMC4251694] [PubMed: 25404155]
- Lipson SJ. Dysplasia of the odontoid process in Morquio's syndrome causing quadriparesis. J Bone Joint Surg Am. 1977;59:340鈥�4. [PubMed: 403192]
- McKay SD, Al-Omari A, Tomlinson LA, Dormans JP. Review of cervical spine anomalies in genetic syndromes. Spine (Phila Pa 1976) 2012;37:E269鈥�77. [PubMed: 22045003]
- Mackenzie WG, Dhawale AA, Demczko MM, Ditro C, Rogers KJ, Bober MB, Campbell JW, Grissom LE. Flexion-extension cervical spine MRI in children with skeletal dysplasia: is it safe and effective? J Pediatr Orthop. 2013;33:91鈥�8. [PubMed: 23232386]
- Montaño AM, Tomatsu S, Gottesman GS, Smith M, Orii T. International Morquio A Registry: clinical manifestation and natural course of Morquio A disease. J Inherit Metab Dis. 2007;30:165鈥�74. [PubMed: 17347914]
- Montaño AM, Tomatsu S, Brusius A, Smith M, Orii T. Growth charts for patients affected with Morquio A disease. Am J Med Genet A. 2008;146A:1286鈥�95. [PubMed: 18412124]
- Morgan KA, Rehman MA, Schwartz RE. Morquio's syndrome and its anaesthetic considerations. Paediatr Anaesth. 2002;12:641鈥�4. [PubMed: 12358664]
- Morquio L (1929) Sur une forme de dystrophie. Bull Soc Pediatr. Paris 27:145-52.
- Morrone A, Caciotti A, Atwood R, Davidson K, Chaoyi D, Francis-Lyon P, Harmatz P, Mealiffe M, et al. Morquio A syndrome-associated mutations: A review of alterations in the GALNS gene and a new locus-specific database. Hum Mutat. 2014;35:1271鈥�9. [PMC free article: PMC4238747] [PubMed: 25137622]
- Muhlebach MS, Wooten W, Muenzer J. Respiratory manifestations in mucopolysaccharidoses. Paediatr Respir Rev. 2011;12:133鈥�8. [PubMed: 21458742]
- Nashed A, Al-Saleh J, Gibbons J, MacLusky I, MacFarlane J, Riekstins A, Clarke J, Narange I. Sleep-related breathing in children with mucopolysaccharidosis. J Inherit Metab Dis. 2009;32:544鈥�50. [PubMed: 19562504]
- Neufeld EF, Muenzer J. The mucopolysaccharidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B, eds. The Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill; 2001:3421-52. Available online.
- Ohto U, Usui K, Ochi T, Yuki K, Satow Y, Shimizu T. Crystal structure of human β-galactosidase: structural basis of Gm1 gangliosidosis and Morquio B diseases. J Biol Chem. 2012;287:1801鈥�12. [PMC free article: PMC3265862] [PubMed: 22128166]
- Onçağ G, Ertan Erdinç AM, Cal E. Multidisciplinary treatment approach of Morquio syndrome (Mucopolysaccharidosis Type IVA). Angle Orthod. 2006;76:335鈥�40. [PubMed: 16539564]
- Pruszczynski B, Mackenzie WG, Rogers K, White KK. Spinal Cord Injury After Extremity Surgery in Children With Thoracic Kyphosis. Clin Orthop Relat Res. 2015;473:3315鈥�20. [PMC free article: PMC4562919] [PubMed: 26242281]
- Ransford AO, Crockard HA, Stevens JM, Modaghegh S. Occipito-atlanto-axial fusion in Morquio-Brailsford syndrome. A ten-year experience. J Bone Joint Surg Br. 1996;78:307鈥�13. [PubMed: 8666648]
- Rivera-Colón Y, Schutsky EK, Kita AZ, Garman SC. The structure of human GALNS reveals the molecular basis for mucopolysaccharidosis IV A. J Mol Biol. 2012;423:736鈥�51. [PMC free article: PMC3472114] [PubMed: 22940367]
- Rodriguez ME, Mackenzie WG, Ditro C, Miller TL, Chidekel A, Shaffer TH. Skeletal dysplasias: evaluation with impulse oscillometry and thoracoabdominal motion analysis. Pediatr Pulmonol. 2010;45:679鈥�86. [PMC free article: PMC3338356] [PubMed: 20575094]
- Ruckenstein MJ, Macdonald RE, Clarke JT, Forte V. The management of otolaryngological problems in the mucopolysaccharidoses: a retrospective review. J Otolaryngol. 1991;20:177鈥�83. [PubMed: 1908026]
- Semenza GL, Pyeritz RE. Respiratory complications of mucopolysaccharide storage disorders. Medicine (Baltimore) 1988;67:209鈥�19. [PubMed: 3134589]
- Simmons MA, Bruce IA, Penney S, Wraith E, Rothera MP. Otorhinolaryngological manifestations of the mucopolysaccharidoses. Int J Pediatr Otorhinolaryngol. 2005;69:589鈥�95. [PubMed: 15850680]
- Solanki GA, Martin KW, Theroux MC, Lampe C, White KK, Shediac R, Lampe CG, Beck M, Mackenzie WG, Hendriksz CJ, Harmatz PR. Spinal involvement in mucopolysaccharidosis IVA (Morquio-Brailsford or Morquio A syndrome):presentation, diagnosis and management. J. Inherit Metab Dis. 2013;36:339鈥�55. [PMC free article: PMC3590412] [PubMed: 23385297]
- Tanpaiboon P (2015) Elosulfase alfa/Vimizim for the treatment of mucopolysaccharidosis IVA (MPS IVA). Expert Rev Endocrinol Metab. Sep 28:569-79.
- Theroux MC, Nerker T, Ditro C, Mackenzie WG. Anesthetic care and perioperative complications of children with Morquio syndrome. Paediatr Anaesth. 2012 Sep;22:901鈥�7. [PubMed: 22738181]
- Tomatsu S, Montaño AM, Nishioka T, Gutierrez MA, Peña OM, Tranda Firescu GG, Lopez P, Yamaguchi S, Noguchi A, Orii T. Mutation and polymorphism spectrum of the GALNS gene in mucopolysaccharidosis IVA (Morquio A). Hum Mutat. 2005;26:500鈥�12. [PubMed: 16287098]
- Tomatsu S, Montaño AM, Oikawa H, Smith M, Barrera L, Chinen Y, Thacker MM, Mackenzie WG, Suzuki Y, Orii T. Mucopolysaccharidosis type IVA (Morquio A disease): clinical review and current treatment. Curr Pharm Biotechnol. 2011;12:931鈥�45. [PubMed: 21506915]
- Tong CK, Chen JC, Cochrane DD. Spinal cord infarction remote from maximal compression in a patient with Morquio syndrome. J Neurosurg Pediatr. 2012 Jun;9:608鈥�12. [PubMed: 22656250]
- Walker PP, Rose E, Williams JG. Upper airways abnormalities and tracheal problems in Morquio's disease. Thorax. 2003;58:458鈥�9. [PMC free article: PMC1746674] [PubMed: 12728175]
- Walker R, Belani KG, Braunlin EA, Bruce IA, Hack H, Harmatz PR, Jones S, Rowe R, Solanki GA, Valdemarsson B. Anaesthesia and airway management in mucopolysaccharidosis. J Inherit Metab Dis. 2013;36:211鈥�9. [PMC free article: PMC3590422] [PubMed: 23197104]
- White KK, Jester A, Bache CE, Harmatz PR, Shediac R, Thacker MM, Mackenzie WG. Orthopedic management of the extremities in patients with Morquio A syndrome. J Child Orthop. 2014;8:295鈥�304. [PMC free article: PMC4128951] [PubMed: 25001525]
- Wilson W, Taubert KA, Gewitz M, Lockhart PB, Baddour LM, Levison M, Bolger A, Cabell CH, Takahashi M, Baltimore RS, Newburger JW, Strom BL, Tani LY, Gerber M, Bonow RO, Pallasch T, Shulman ST, Rowley AH, Burns JC, Ferrieri P, Gardner T, Goff D, Durack DT., American Heart Association Rheumatic Fever, Endocarditis, and Kawasaki Disease Committee. American Heart Association Council on Cardiovascular Disease in the Young; American Heart Association Council on Clinical Cardiology; American Heart Association Council on Cardiovascular Surgery and Anesthesia; Quality of Care and Outcomes Research Interdisciplinary Working Group. Prevention of infective endocarditis: guidelines from the American Heart Association: a guideline from the American Heart Association Rheumatic Fever, Endocarditis, and Kawasaki Disease Committee, Council on Cardiovascular Disease in the Young, and the Council on Clinical Cardiology, Council on Cardiovascular Surgery and Anesthesia, and the Quality of Care and Outcomes Research Interdisciplinary Working Group. Circulation. 2007;116:1736鈥�54. [PubMed: 17446442]
- Wood TC, Harvey K, Beck M, Burin MG, Chien YH, Church HJ, D'Almeida V, van Diggelen OP, Fietz M, Giugliani R, Harmatz P, Hawley SM, Hwu WL, Ketteridge D, Lukacs Z, Miller N, Pasquali M, Schenone A, Thompson JN, Tylee K, Yu C, Hendriksz CJ. Diagnosing mucopolysaccharidosis IVA. J Inherit Metab Dis. 2013;36:293鈥�307. [PMC free article: PMC3590423] [PubMed: 23371450]
- Wraith JE. The mucopolysaccharidoses: a clinical review and guide to management. Arch Dis Child. 1995;72:263鈥�7. [PMC free article: PMC1511064] [PubMed: 7741581]
- Yamada N, Fukuda S, Tomatsu S, Muller V, Hopwood JJ, Nelson J, Kato Z, Yamagishi A, Sukegawa K, Kondo N, Orii T. Molecular heterogeneity in mucopolysaccharidosis IVA in Australia and Northern Ireland: nine novel mutations including T312S, a common allele that confers a mild phenotype. Hum Mutat. 1998;11:202鈥�8. [PubMed: 9521421]
Chapter Notes
Author Notes
Dr. Oetgen, an attending surgeon in the Department of Orthopaedics and Sports Medicine at Children’s National Medical Center, has a special interest in the orthopaedic manifestations of genetic conditions affecting pediatric patients, including the mucopolysaccharidoses. He participates in the care of these patients in collaboration with the Children’s National Medical Center Department of Genetics.
Dr. Tanpaiboon is an attending physician in the Division of Genetics and Metabolism at Children’s National Medical Center. Her main interest is the field of Inborn Errors of Metabolism, particularly the lysosomal storage disorders (LSDs). She has been actively involved in international multicenter clinical trials of enzyme replacement therapy for MPS IVA and other LSDs.
Revision History
- 24 March 2016 (ma) Comprehensive update posted live
- 13 March 2014 (pt) Revision: information on enzyme replacement therapy and MRI added
- 11 July 2013 (me) Review posted live
- 18 January 2013 (pt) Original Submission