
摘要
临床特征
Alström 综合征是临床主要表现为锥杆营养不良,肥胖,进行性感觉神经性听力减退,扩张型或限制型心肌病和胰岛素抵抗的综合征,并伴随多器官衰竭。患者临床表现多 样,即便在同一家系中都有很高的表型可变性。锥杆营养不良临床表现为进行性视力减退,畏光和眼球震颤,这些症状通常于出生到15个月间就会出现。很多患者 在20岁彻底失去光感,但部分患者可保持大字体的阅读能力至30岁。患儿常于出生时体重正常,但在出生后一年发展为躯干性肥胖。70%以上的患者在出生后 一年内表现出进行性听力减退。听力减退可在10岁至20岁发展至中等或严重水平(40-70db)。胰岛素抵抗都会伴随黑棘皮症,并于30岁左右发展为2 型糖尿病。几乎所有的患者都表现出血脂异常。其他内分泌系统异常包括甲状腺功能减退,男孩的低促性腺素型功能减退症和女孩的多囊卵巢。
超 过 60%的Alström综合征患者会出现由于扩张型和限制型心肌炎所引发的心衰。50%的患者早期发育里程碑推迟,智力正常。肝脏异常表现为转氨酶水平升 高,脂肪变性,肝脾肿大和脂肪肝。高血压和肝硬化可导致肝性脑病和威胁生命的食管静脉曲张。另外也可能会出现肺功能紊乱和严重的肾病。青少年末期可能会发 展至终末期肾病。
诊断/检测
Alström综合征的诊断主要依靠临床所见。ALMS1基因是Alström 综合征目前所知的唯一致病基因,在70%-80%的北欧裔患者中可检出ALSM1基因的致病突变,而检出率在全世界范围内大约为40%。管理
Management.
对症治疗 :可针对畏光配红色处方眼镜,做盲目或视力减退方面的引导;健康饮食和规律运动;当出现听力下降时行鼓膜切开置管术或佩戴助听器。对 心肌病需采取抗充血措施;对于II型胰岛素拮抗型糖尿病采取常规治疗措施。高脂血采用常规的尼克酸类药物。如果青春性发育不正常需咨询内分泌专家。对排泄 困难的患者需施尿流改道术或导管术,已有一些患者被报道成功进行了肾移植手术。对门静脉高压和食管静脉曲张需采取合理的治疗方案,慢性阻塞性肺病和相关的 感染需根据业内公认指南进行治疗。
主要并发症的预防:进行性糖尿病和严重的高血糖症需通过正确的生活方式和避免过度肥胖来控制
次要并发症的预防:常规的小儿科免疫接种,在急性病和手术中需实时监控心脏状态和供氧。
检 测:年度检查视力和听力;体重,身高和BMI指数;心脏状态(包括超声心动图);血浆胰岛素浓度;血脂分析;ALT,AST和GGT浓度检测;肺功能;甲 状腺功能。每两个月到三个月检测空腹血糖浓度;密切关注空腹和餐后血糖浓度的升高水平。两年一次验尿和血液中电解质、尿酸、尿氨素浓度。如尿检或出现相关 症状,需及时进行肾脏和膀胱的超声检查。需避免的药剂和环境因素:任何肾功能和心功能不全患者禁用药物;可能影响其他系统的对症治疗方案(如针对糖尿病的二苯硫卡巴腙对于心功能不全的患者是禁用的)
遗传咨询
Alström 综合征是常染色体隐性遗传病,患者的兄弟姐妹有25%的可能性是患者,50%的可能性是无症状的携带者,25%的可能性是无症状的非携带者。如果在家系中已有人确定是Alstrom综合征患者,那么建议对家系中有血缘关系的成员进行携带者筛查和产前诊断。
诊断
临床诊断
Alstrom综合征的诊断是依据婴儿期、儿童期和青年期出现的主要临床特点而做的 (见表1和图1).
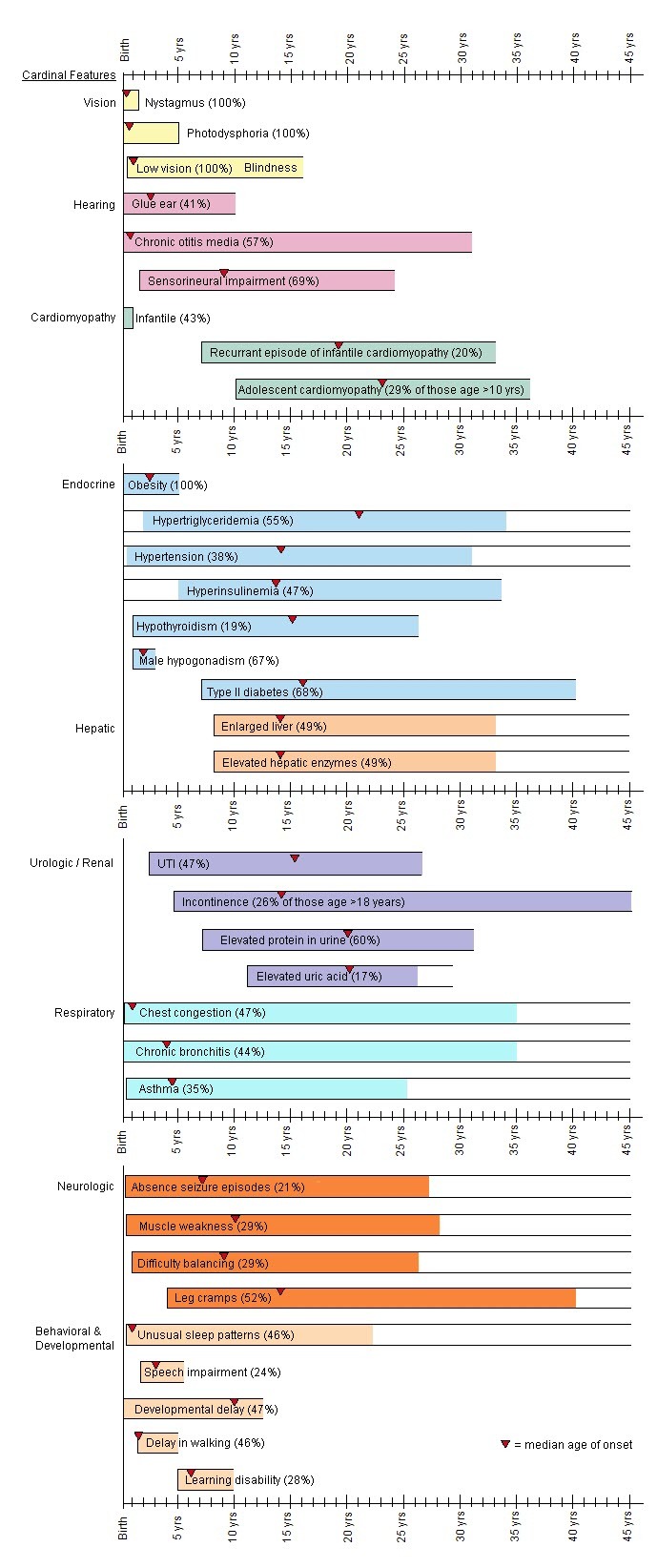
图1.
Alstrom综合征不同年龄段的临床特点
患者不同年龄阶段的主要临床特点[Marshall et al 2005]:
主要特点
- 视锥杆营养不良,伴眼球震颤和畏光(光敏感/畏光)常发作于出生后第一年
- 肥胖,躯干型肥胖(BMI>25,或大于95%百分位),常出现于儿童期早期
- 进行性双边型感觉神经性耳聋在1~10岁出现,发病年龄多样。听力减退主要从高频段开始
- 扩张型心肌病于婴儿期发作或60%左右的患者于青年期或成年期发作限制型心肌病
- 肺病的症状从频繁发作的支气管感染至肺纤维化和肺动脉高压
- 胰岛素耐受/2型糖尿病,胰岛素耐受程度根据个体差异从高胰岛素血症到葡萄糖不耐受到2型糖尿病不等。2型糖尿病可能从儿童期到青年期发病。
- 肝脏方面的症状多变,从转氨酶升高到脂肪性肝炎。肝脾可能会出现扩大症状。广泛型肝硬化,纤维化,门静脉高压和肾衰都有案例被报道。
- 肾脏方面的表型是进行性的,肾小球硬化的严重程度高度可变
其他特点
- 甲状腺功能减退
- 牙齿异常(如牙釉质变色,牙位异常或额外牙)
- 宽平足
- 男性性腺功能低下症/女性雄激素过多症
- 泌尿道功能紊乱/逼尿肌不稳定
- 发育里程碑延迟,包括大运动延迟和语言表达能力延迟
- MRI异常信号(部分患者蝶鞍缺失),强直阵挛发作
- 面容异常(下置耳,圆脸,额叶内板增生,厚耳,额过早秃顶,毛发细)
据 Marshall et al [2007a]报道,诊断标准根据患者的年龄进行修订 (表 1).
Table 1.
不同年龄的诊断标准
| 年龄范围 | 诊断标准 | 判断标准 | 其他可变支持证据 | |
|---|---|---|---|---|
| 主要 | 次要 | |||
| 出生-2岁1 |
|
|
2个主要证据或1个主要证据+2个次要证据 |
|
| 3-14 岁 |
|
|
2个主要证据或1个主要证据+3个次要证据 |
|
| 15岁-成年 |
|
|
2个主要证据+2个次要证据活1个主要证据+4个次要证据 |
|
Adapted from Marshall et al [2007a]. Reprinted with permission of Nature Publishing Group
Note: The diagnosis is established in individuals of all ages in whom two ALMS1 pathogenic variants are identified.
ERG = electroretinogram
T2DM = type 2 diabetes mellitus
DCM/CHF = dilated cardiomyopathy with congestive heart failure
UTI = urinary tract infections
1.
Children in this age group should be reevaluated for the presence of major and minor criteria after infancy.
Molecular Genetic Testing
Gene.ALMS1 is the only gene in which mutation is known to cause Alström syndrome.
Clinical testing
Table 2.
Summary of Molecular Genetic Testing Used in Alström Syndrome
| Gene 1 | Test Method | Variants Detected 2 | Variant Detection Frequency by Test Method 3 |
|---|---|---|---|
| ALMS1 | Targeted analysis for pathogenic variants | 19-bp insertionexon 16 4 | See footnote 4 |
| Sequence analysis 5 of select exons: 16, 10, and 8 6 | Sequence variants | 25%-40% 7, 8 | |
| Sequence analysis 5 of entire coding region 9 | Sequence variants | Two mutated alleles in 8/12 individuals 8, 10 | |
| Deletion/duplication analysis 11 | Exon and whole-gene deletions | Unknown 8, 12 |
See Table A. Genes and Databases for chromosomelocus and protein.
2.
See Molecular Genetics for information on allelic variants.
3.
The ability of the test method used to detect a variant that is present in the indicated gene
4.
Identifed in Acadian kindred [Collin et al 2002]
5.
Examples of pathogenic variants detected by sequence analysis may include small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exon or whole-gene deletions/duplications are not detected. For issues to consider in interpretation of sequence analysis results, click here.
6.
Exons sequenced may vary by laboratory.
7.
8.
Although there is no evidence for locus heterogeneity in Alström syndrome, other possible explanations for the low variant detection frequency are that: (1) sequence analysis of select exons misses pathogenic variants elsewhere in gene; (2) deletion of an exon(s) would not be detected by sequence analysis; and (3) diagnostic confusion may exist between Alström syndrome and other disorders such as Bardet-Biedl syndrome (BBS) (see Differential Diagnosis).
9.
Newer sequencing methodologies have also been applied [Bell et al 2011, Pereiro et al 2011].
10.
In a study of 12 individuals from the UK in which Minton et al [2006] sequenced the entire coding region, both pathogenic variants were identified in 8/12, one pathogenic variant was identified in 2/12, and no variants were identified in 2/12.
11.
Testing that identifies deletions/duplications not readily detectable by sequence analysis of the coding and flanking intronic regions of genomic DNA; included in the variety of methods that may be used are: quantitative PCR, long-range PCR, multiplex ligation-dependent probe amplification (MLPA), and chromosomal microarray (CMA) that includes this gene/chromosome segment.
12.
A gross deletion has been reported in a consanguineous family from northern Pakistan [Bond et al 2005].
Test characteristics. See Clinical Utility Gene Card [Marshall et al 2011a] for information on test characteristics including sensitivity and specificity.
Interpretation of test results. Given the current detection rate, failure to identify a disease-causing sequence variant does not preclude the diagnosis of Alström syndrome.
Testing Strategy
To confirm/establish the diagnosis in a proband. The diagnosis of Alström syndrome relies primarily on clinical findings and/or family history.
- In some instances the diagnosis can be confirmed by molecular genetic testing. Sequence analysis of the coding region should be performed: tiered testing with sequencing of select exons first, followed by analysis of the entire gene.
- The frequency of deletions is unknown but deletion/duplication analysis may be clinically indicated in some instances.
Carrier testing for at-risk relatives requires prior identification of the pathogenic variants in the family.
Note: Carriers are heterozygotes for this autosomal recessive disorder and are not at risk of developing the disorder.
Prenatal diagnosis and preimplantation genetic diagnosis (PGD) for at-risk pregnancies require prior identification of the pathogenic variants in the family.
Clinical Characteristics
Clinical Description
A wide range of clinical variability is observed among individuals with Alström syndrome, including among sibs [Hoffman et al 2005]. The first clinical presentation of Alström syndrome (Table 3) is usually nystagmus caused by cone-rod dystrophy and resulting in childhood blindness. Disease characteristics that are later in onset include truncal obesity that manifests during the first year of life, progressive sensorineural hearing loss, infantile-onset dilated cardiomyopathy or later-onset restrictive cardiomyopathy, insulin-resistant type 2 diabetes mellitus, and hepatic, pulmonary, and renal dysfunction.
Table 3.
Age of Onset and Incidence of Common Features of Alström Syndrome
| Feature | Age of Onset Range (Mean) | Incidence |
|---|---|---|
| Cone-rod dystrophy | Birth - 15 mos (5 mos) | 100% |
| Obesity | Birth - 5 years (2.5 yrs) | 98% |
| Progressive sensorineural hearing loss | 2-25 yrs (9 yrs) | 88% |
| Dilated cardiomyopathy | 2 wks - 4 mos | 42% |
| Restrictive cardiomyopathy | Juvenile - late 30s | 18% |
| Insulin resistance / type 2 diabetes mellitus | 4-30 yrs / 8-40 yrs (16 yrs) | 92% / 68% |
| Developmental delay | Birth-adolescence | 25%-30% |
| Short stature | Puberty - adult | 98% |
| Hypogonadotropic hypogonadism | 10+ yrs | 78% of males |
| Urologic disease | Adolescence - adult | 48% |
| Renal disease | Adolescence - adult | Variably progressive with age in all individuals |
| Hepatic disease | 8-30 yrs | 23%-92% |
Based on a study of 182 patients by Marshall et al [2005]
Cone-rod dystrophy. In most affected individuals, visual problems present as progressive cone dystrophy resulting in visual impairment, photophobia, and nystagmus between birth and age 15 months. Full-field electroretinography (ERG), required to establish the diagnosis of cone-rod dystrophy, is abnormal from birth, eventually with impairment of both cone and rod function. Rod function is preserved initially but deteriorates as the individual ages, with visual acuity of 6/60 or less by age ten years, increasing constriction of visual fields, and no light perception by age 20 years. In some rare cases, some vision (e.g., the ability to read large print) is retained into the third decade. The severity and age of onset of the retinal degeneration vary among affected individuals [Malm et al 2008].
Fundus examination in the first decade may be normal or may show a pale optic disc and narrowing of the retinal vessels. Posterior subcapsular cataracts are common, even in the absence of diabetes.
Obesity. Children with Alström syndrome have normal birth weight. Hyperphagia and excessive weight gain (some studies show weight gain greater than expected for intake) begin during their first year, resulting in childhood truncal obesity. In some individuals body weight tends to normalize, decreasing into the high-normal to normal range after adolescence. The degree of obesity may mirror cultural background, with higher levels in North America than in South America, Turkey, Japan, and parts of Europe [Koç et al 2006].
Progressive bilateral sensorineural hearing loss. Hearing loss may be detected as early as age one year, initially in the high frequency range. Progressive sensorineural hearing loss presents in the first decade in as many as 70% of individuals with Alström syndrome. Hearing loss may progress to the severe or moderately severe range (40-70 db) by the end of the first to second decade [Van den Abeele et al 2001]. A high incidence of glue ear (long-standing sticky fluid in the middle ear) can lead to an additional conductive hearing loss [Marshall et al 2005].
Cardiomyopathy. More than 60% of individuals with Alström syndrome develop congestive heart failure as a result of cardiomyopathy at some stage of their lives. Onset, progression, and clinical outcome of the cardiomyopathy vary, even within families [Makaryus et al 2003, Hoffman et al 2005].
More than 40% of affected infants have a transient but severe dilated cardiomyopathy with onset between age three weeks and four months [Marshall et al 2005]. Most of these children survive and make an apparently full recovery in infancy. At a later age about 10%-15% of this group have spontaneous recurrence of a progressive restrictive cardiomyopathy. The proportion of those with Alström syndrome who develop infantile-onset cardiomyopathy may be underestimated because some infants who succumb may have undiagnosed Alström syndrome.
About 20% of individuals with Alström syndrome have later-onset progressive restrictive cardiomyopathy identified between the teens to late 30s. Postmortem myocardial fibrosis has been described [Marshall et al 2005]. Cardiac magnetic resonance imaging suggests that myocardial fibrosis may be present in clinically affected and asymptomatic individuals [Loudon et al 2009].
Insulin resistance/type 2 diabetes mellitus. Diabetes mellitus in Alström syndrome is the result of tissue resistance to the actions of insulin, as demonstrated by an elevated plasma insulin concentration and glucose intolerance that usually present in childhood. The age at which type 2 diabetes mellitus develops varies; it may be as early as age five years. Type 2 diabetes mellitus and insulin resistance are typically accompanied by the skin changes of acanthosis nigricans, i.e., velvety hyperpigmented patches in intertriginous areas.
In a small study of 12 unrelated individuals with Alström syndrome, obesity (BMI and waist circumference) decreased with age, whereas insulin resistance increased with age [Minton et al 2006].
Coronary artery disease as a result of insulin resistance, diabetes, dyslipidemia, and renal failure has been reported in one affected individual [Jatti et al 2012]. Diabetic peripheral neuropathy with risk of foot ulceration (which would be expected to develop) has been found to occur rarely if at all [Paisey et al 2009].
Hyperlipidemia. Hyperlipidemia is primarily hypertriglyceridemia, but can sometimes include high serum concentration of total cholesterol. Affected individuals are at risk for sudden increase in triglycerides precipitating life-threatening pancreatitis [Wu et al 2003].
Developmental delay. About 20% of affected individuals have delay in early developmental milestones including delays in gross and fine motor skills and in expressive and receptive language. About 30% have a learning disability. Cognitive impairment (IQ <70) is very rare.
Short stature. Growth rates for young children are normal, but accelerated skeletal maturity (2-3 years advanced bone age) and low-serum growth hormone concentrations result in adult stature that is typically less than the 25th centile. In about 98% of individuals over age 16 years height is below the fifth centile [Maffei et al 2007].
Scoliosis or kyphosis, beginning in puberty, is common [Maffei et al 2002].
Male pubertal development. There is a variable mixture of hypogonadotropic hypogonadism and testicular fibrosis which can result in delayed or arrested puberty. As a result secondary sexual characteristics may be normal or immature; gynecomastia may be present [Marshall et al 2005]. This mixed causality has meant that puberty is induced and androgenization maintained with testosterone rather than gonadotropins. Male fertility has not been systematically studied in the syndrome but has not been reported in an affected individual with pathogenic variants in both copies of ALMS1.
Female pubertal development. Endocrine disturbances in females include reduced plasma gonadotropin concentrations, hirsutism, polycystic ovarian syndrome consistent with insulin resistance, precocious puberty (pubertal onset before age 8 years), endometriosis, irregular menses, or amenorrhea. External genitalia are normal in females, though breast development is often poor. Fertility in females has not been systematically studied. Although a molecular diagnosis was not confirmed, one clinical report describes two unrelated females with late presentation of the syndrome, each of whom had healthy children [Iannello et al 2004].
Urologic disease. Urinary problems affect approximately 50% of individuals with Alström syndrome. Urologic disorders of varying severity, characterized by detrusor-urethral dyssynergia (lack of coordination of bladder and urethral muscle activity), have been described. The greatest problems appear to occur in females in their late teens. Minor symptoms include urgency and long intervals between voiding, which suggests a decrease in bladder sensation, hesitancy, and poor urinary flow. Moderate symptoms include urinary frequency, incontinence, and symptoms associated with recurrent infections. More severe urinary symptoms include worsening urinary incontinence or retention; these symptoms may alternate. Lower abdominal and perineal pain is common and may relate to abnormal bladder/sphincter function [MacDermott 2001]. Intermittent self-catheterization has been helpful in a small number of affected individuals and cystectomy with an ileal conduit has been performed in isolated severe cases.
Renal disease is common, slowly progressive, and highly variable, and may be unrelated to diabetes; it manifests as tubulo-interstitial disease resulting from interstitial fibrosis. Initial signs of mildly elevated serum concentrations of creatinine and blood urea nitrogen (BUN) may be followed by polyuria and polydipsia resulting from a concentrating defect. Onset can be in mid-childhood through adulthood. End-stage renal disease (ESRD) can occur as early as the mid- to late teens.
Renal biopsy often shows interstitial fibrosis, glomerular hyalinosis, and tubular atrophy [Marshall et al 2005].
Obstructive uropathy is rare.
Hepatic disease. Elevated plasma concentration of liver enzymes is common in early childhood. Macrovesicular steatosis and hepatomegaly can be present or absent. Progression to cirrhosis and hepatic failure can occur in the second to third decades, and the complications of portal hypertension, ascites, splenomegaly, hepatic encephalopathy, and esophageal varices are also seen.
Liver biopsies and postmortem examination have revealed varying degrees of steatohepatitis, hepatic fibrosis, cirrhosis, chronic nonspecific active hepatitis with lymphocytic infiltration, patchy necrosis, and fatty liver [Quiros-Tejeira et al 2001, Marshall et al 2005].
Gastrointestinal disease. General GI disturbances such as epigastric pain and gastroesophageal reflux disease (GERD) are common. Cecal volvulus without adhesions or obstructive lesions was reported in a sibling pair, raising the possibility of a generalized smooth muscle dysfunction in the syndrome [Khoo et al 2009].
Pulmonary involvement. Recurrent chest infections are common at all ages, but establishment of COPD is problematic because of difficulty in accurate performance of spirometry. Most frequently there is a combination of restrictive lung disease due to kyphoscoliosis and sometimes pulmonary fibrosis, which has been confirmed in post mortem tissue and may be detected in life with high-resolution chest CT scan. This may progress (with the added effects of cardiomyopathy) to pulmonary hypertension. The resulting susceptibility to severe hypoxia postoperatively or during episodes of pneumonia has been reported [Khoo et al 2009, Florentzson et al 2010].
Other
- Scoliosis and kyphosis of varying severity are reported in 30%-70% of individuals with Alström syndrome [Van den Abeele et al 2001, Marshall et al 2005].
- Severe flat feet and dental abnormalities have been observed [Koray et al 2001, Marshall et al 2005].
- Hypothyroidism has been reported in nearly 20% of individuals [Marshall et al 2005]; hyperthyroidism occurs infrequently [Ozgül et al 2007].
- Hypertension, often beginning in childhood, has been described in 30% of individuals [Marshall et al 2005].
- Neurobehavioral manifestations such as absence seizures, excessive startle response, severe and unexplained peripheral pain, and mild autistic spectrum behaviors have been reported [Marshall et al 2005].
Genotype-Phenotype Correlations
In one study involving fewer than 12 kindreds, no correlations were found between pathogenic variant position and the occurrence of specific clinical features, such as dilated cardiomyopathy [Minton et al 2006].
A separate genotype-phenotype association study of 58 affected individuals [Marshall et al 2007b] found suggestive associations between disease-causing variants in exon 16 of ALMS1 and the following findings:
- Onset of retinal degeneration before age one year (P=0.02)
AND - Occurrence of urologic dysfunction (P=0.02), diabetes mellitus (P=0.03), and dilated cardiomyopathy (P=0.03)
A more significant association was found between alterations in exon 8 and absent, mild, or delayed renal disease (P=0.0007).
This preliminary observation may have implications for the understanding of how alternative splicing of ALMS1 contributes to the severity of the disease, and thus provide useful information to clinicians.
Penetrance
No clinically unaffected individuals with two ALMS1 pathogenic variants in trans have been reported; thus, penetrance appears to be 100% [Marshall et al 2011a].
Prevalence
The prevalence of Alström syndrome is difficult to estimate; it is likely underdiagnosed because of its rarity. Estimates of the prevalence range from 1:10,000 [Minton et al 2006] to fewer than 1:1,000,000 [Marshall et al 2011b, Orphanet].
About 800 individuals diagnosed with Alström syndrome have been identified worldwide.
Ethnically or geographically isolated populations have a higher-than-average frequency of Alström syndrome [Deeble et al 2000, Ozgül et al 2007, Aldahmesh et al 2009].
Genetically Related (Allelic) Disorders
No other phenotypes are known to be caused by pathogenic variants in ALMS1.
Differential Diagnosis
Bardet-Biedl syndrome (BBS) shares some features of Alström syndrome. The major clinical features of BBS are rod-cone dystrophy, postaxial polydactyly, central obesity, cognitive impairment, hypogonadism, and renal dysfunction [Goldstone & Beales 2008]. A major difference between Alström syndrome and BBS is the timing of the onset of visual problems: in Alström syndrome, visual problems are usually apparent in the first two years of life; in BBS, the average age of onset of visual problems is 8.5 years. Polydactyly, which is common in BBS, has not been described in Alström syndrome. Cognitive impairment is well described in BBS, while in most persons with Alström syndrome intelligence is normal, but delays in early milestones have been reported. Other differences include the relative infrequency of hearing problems (~5%) and diabetes mellitus (5%-10%) in BBS compared with Alström syndrome. Pathogenic variants in at least 14 different genes are causative. Inheritance is autosomal recessive.
Achromatopsia, a disorder that affects only the retina, is characterized by reduced visual acuity, pendular nystagmus, increased sensitivity to light (photophobia), a small central scotoma, eccentric fixation, and reduced or complete loss of color discrimination. Most individuals have complete achromatopsia with total lack of function of all three types of cones (i.e., the long-wavelength-sensitive cones [red], the middle-wavelength-sensitive cones [green] and the short-wavelength-sensitive cones [blue]). Rarely, individuals have incomplete achromatopsia, in which one or more cone types may be partially functioning resulting in symptoms similar to but less severe than those of complete achromatopsia. Nystagmus and increased sensitivity to bright light develop shortly after birth. Best visual acuity ranges from 20/200 or less in complete achromatopsia to as high as 20/80 in incomplete achromatopsia. Visual acuity is usually stable over time. The diagnosis of achromatopsia is based on case history, color vision testing, electrophysiologic examination, and absent or only minor fundus changes. The fundus is usually normal. Pathogenic variants in three genes, CNGA3, CNGB3, and GNAT2, are causative. Inheritance is autosomal recessive.
Leber congenital amaurosis(LCA), a severe dystrophy of the retina without other organ system involvement, typically becomes evident in the first year of life. Reduced vision is accompanied by nystagmus, sluggish pupillary responses, photophobia, hyperopia, and keratoconus. The electroretinogram (ERG) is characteristically "nondetectable" or severely subnormal. The oculo-digital sign (repeated eye rubbing, poking, and pressing of the eyes) is characteristic. Although the retina may appear normal in infancy, a pigmentary retinopathy reminiscent of retinitis pigmentosa is frequently observed later in childhood. The 15 genes currently known to be associated with LCA are: GUCY2D (locus name: LCA1), RPE65 (LCA2), SPATA7 (LCA3), AIPL1 (LCA4), LCA5 (LCA5), RPGRIP1 (LCA6), CRX (LCA7), CRB1 (LCA8), CEP290 (LCA10), IMPDH1 (LCA11), RD3 (LCA12), RDH12 (LCA13), LRAT (LCA14), TULP1 (LCA15) and KCNJ13 (LCA16) . Depending on the survey, these genes together are estimated to account for from one third to one half of all LCA. Three other disease loci for LCA have been reported. Most often, Leber congenital amaurosis is inherited in an autosomal recessive manner. Rarely, LCA is inherited in an autosomal dominant manner as a result of pathogenic variants in CRX.
Early-onset dilated cardiomyopathy.Dilated cardiomyopathy, characterized by cardiac dilation and reduced systolic function, is the end stage of a number of inherited and acquired disorders. Familial dilated cardiomyopathy may be inherited in an autosomal dominant manner and less frequently in an autosomal recessive manner with ventricular dilatation and systolic dysfunction becoming apparent in the third and fourth decades.
Inherited mitochondrial disorders represent a heterogeneous group of complex disorders that may be caused by pathogenic variants in mitochondrial DNA or nuclear DNA. Clinical features common to mitochondrial disorders and Alström syndrome include cardiomyopathy, sensorineural deafness, optic atrophy, pigmentary retinopathy, and diabetes mellitus; however, central nervous system involvement and muscle weakness occur in individuals with mitochondrial disorders, while they are not reported in Alström syndrome. Generally, mitochondrial disorders present in late childhood or in adulthood, unlike Alström syndrome, which usually presents during the first year of life.
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease in an individual diagnosed with Alström syndrome, a complete history of disease should direct detailed physical examination and investigations. A multidisciplinary team should be established to formulate and coordinate monitoring, management, and therapeutic interventions.
- Cone-rod dystrophy. Ophthalmologic evaluation, including electroretinogram and visual field testing
- Obesity. Measurement of weight and height; calculation of body mass index (BMI)
- Progressive sensorineural hearing loss. Audiometry with auditory brain stem response (ABR) and otoacoustic emissions (OAE) to detect sensorineural or conductive hearing loss; assessment of otitis media and conductive hearing loss
- Dilated cardiomyopathy. A detailed cardiac history and examination, including auscultation, echocardiography and ECGs. Echocardiography is required to demonstrate ventricular dilation, fibrosis, and decreased myocardial function.
- Insulin resistance/type 2 diabetes mellitus
- Fasting plasma glucose, even in infancy
- A glucose tolerance test (GTT) as early as age six years
- Plasma insulin concentration, as hyperinsulinemia may be present from infancy
- Hyperlipidemia. A fasting lipid profile, including triglycerides
- Endocrine abnormalities
- Assessment of thyroid function, including plasma TSH and free T4 concentration. If hyperthyroidism is suspected, free T3 concentration should be done.
- Measurement of pituitary hormones
- Urologic
- History to document urinary difficulties
- If the individual is symptomatic or if urinalysis is abnormal, renal ultrasound examination to detect pelvi-calyceal dilatation and bladder ultrasound examination to measure post-voiding residual volumes
- Renal disease
- Baseline blood pressure; 24-hour blood pressure monitoring
- Measurement of plasma BUN, creatinine, urea and electrolytes. If renal function testing is abnormal, ultrasound examination. If abnormalities, refer to a nephrologist.
- Hepatic disease
- Measurement of plasma ALT, AST, and GGT concentration
- Liver ultrasonography to evaluate for possible hepatomegaly and portal hypertension
- If clinically indicated, screening esophagogastroduodenoscopy for varices
- Pulmonary disease. Detailed assessment of pulmonary function by chest radiography, combined with pulmonary function tests
- Developmental assessment. Educational evaluation for intervention and IEP
- Other
- Gastrointestinal. If symptoms of reflux esophagitis remain severe, despite acid blockers, perform barium swallow or upper gastrointestinal endoscopy.
- Skin. Note acanthosis nigricans (indication of insulin resistance/diabetes mellitus), alopecia, body hair, hirsutism on physical examination.
- Orthopedic abnormalities. Note flat feet, scoliosis, barrel chest, kyphoscoliosis on physical examination.
- Neurologic manifestations. Neurologic evaluation with EEG to examine for seizures. Note autistic-spectrum behavioral abnormalities, excessive startle, tactile defensiveness, unexplained joint or muscle pain, muscle dystonia, or hyporeflexia.
- Medical genetics consultation
Treatment of Manifestations
No therapy exists to prevent the progressive organ involvement. However, monitoring for developing problems and early intervention is essential.
Rod-cone dystrophy
- Early on when photodysphoria is significant, the use of red-orange tinted prescription lenses may reduce symptoms.
- Early educational planning should be based on the certainty of blindness. Instruction in the use of Braille, mobility training, adaptive living skills, and computing skills (including voice recognition and transcription software), and the use of large print reading materials while vision is still present are crucial.
Obesity. A healthful, reduced calorie diet and regular exercise, such as walking, hiking, biking, and swimming with adaptations for the blind, are recommended to control weight gain.
Progressive sensorineural hearing loss
- Myringotomy has been helpful in individuals with recurrent otitis media (‘glue ear’).
- Hearing can be maximized with bilateral digital hearing aids.
- Cochlear implantation has benefitted some patients [Florentzson et al 2010].
Cardiomyopathy. Angiotensinogen-converting enzyme (ACE) inhibitors, diuretics, digoxin, and possibly beta-blockers should be used in the treatment of cardiac failure. Cardiac transplantation has been successful in isolated cases [Goerler et al 2007].
Insulin resistance/type 2 diabetes should be treated as in the general population unless heart failure and/or liver dysfunction are present. The diabetes mellitus is characterized by insulin resistance, but some individuals respond to a low-sugar, low-fat diet; exercise; and metformin. Glitazones are added to further reduce insulin resistance but must be avoided in the presence of active or treated heart failure. These treatments should be discontinued when the serum creatinine concentration exceeds 200 µmol/L or if cardiomyopathy is evident. Incretin analogues given subcutaneously, as in nonsyndromic type 2 diabetes, are successful in two thirds of cases [Paisey et al 2008, Paisey 2009].
Hypertriglyceridemia
- Nicotinic acid derivatives can be helpful in long-term reduction of severe hypertriglyceridemia (>20 mmol/l) especially if pancreatitis has occurred and diabetes is absent or well controlled [Paisey et al 2004, Paisey 2009]. Statins are unlikely to be effective but can be considered for long-term prevention of atherosclerosis in adults with low HDL, high LDL, and diabetes.
- Pancreatitis should be treated as in the general population.
Endocrine
- As children approach puberty, gonadotropin and pituitary hormones should be assessed to determine if hormonal adjustments are necessary.
- Male hypogonadism should be treated with testosterone according to local endocrine guidelines to preserve sexuality, muscle strength, and bone health.
- Thyroxine therapy should be initiated and monitored if the individual is hypothyroid.
Urologic. Some individuals have required urinary diversion or self-catheterization to manage voiding difficulties [MacDermott 2001].
Renal disease
- The use of enzyme ACE inhibitors may be considered if proteinuria is detected.
- Successful renal transplantation has occurred in a growing number of individuals, but can be contraindicated in the presence of other complications including morbid obesity, uncontrolled diabetes, and cardiomyopathy.
Hepatic disease. Portal hypertension may be treated with beta-blockade and sclerotherapy of the esophageal veins. Banding should be done in order to prevent upper-GI hemorrhage from varices. Patients who fail to respond to medication and banding are candidates for a transjugular intrahepatic portosystemic shunt (TIPS) to decrease risk of variceal bleeding caused by portal hypertension. Patients with significant portal hypertension should be evaluated early for liver transplantation.
Pulmonary disease. General activity, including breathing exercises, can reduce chronic hypoxia and improve wellbeing. Coaching by an exercise expert may be necessary in order to achieve adequate spirometry results to determine the presence of obstructive airways disease.
Other
- If skeletal abnormalities are present, referral to an orthopedist is appropriate.
- Reflux esophagitis, skin manifestations, orthopedic abnormalities, and neurologic manifestations should be treated as in the general population.
- Education intervention, as indicated by evaluation and IEP (individual education plan) with the expectation of total blindness and hearing loss.
Prevention of Primary Complications
The progression to diabetes mellitus and the severity of hyperglycemia can in some cases be mitigated by lifestyle changes and avoidance of severe obesity.
Prevention of Secondary Complications
Routine pediatric immunizations should be given and administration of pneumococcal vaccination should be considered.
Care must be taken during sedation or operative procedures. The combination of dilated cardiomyopathy, congestive heart failure, pulmonary hypertension, and pulmonary fibrosis can cause sudden severe hypoxia in an affected individual following surgery or even during a minor infection. Close monitoring of cardiac status and oxygenation are necessary until the individual is fully recovered.
Surveillance
Rod-cone dystrophy. Annual ophthalmologic follow-up is indicated as long as the affected individual has vision.
Obesity. Weight, height, and body mass index (BMI) should be recorded annually and plotted on growth curves.
Progressive sensorineural hearing loss. Audiometry should be performed yearly.
Cardiomyopathy
- A detailed cardiac history and examination including echocardiography annually even in the absence of symptoms related to left ventricular dysfunction (signs of cardiac failure, such as sweating, fatigue, lethargy, asthma, decreased physical activity)
- ECGs in parallel with echocardiography and 24-hour ECG monitoring if indicated
- In individuals who have had infantile cardiomyopathy, annual monitoring by a pediatric cardiologist, even if the individual has recovered from cardiomyopathy and is asymptomatic
Insulin resistance/type 2 diabetes
- Annual measurement of plasma insulin concentration, as hyperinsulinemia may be present from early infancy
- Measurement of fasting plasma glucose concentration every two to three months
- If fasting blood glucose is greater than 7 mmol/L, or postprandial blood glucose is greater than 11 mmol/L, measurement of HbA1c concentration and serum glucose concentration regularly (every 6 months, although glucose estimations may be performed more frequently as determined by the 'diabetic control' of the affected individual)
Hyperlipidemia. Annual total lipid profile determination is appropriate unless hyperlipidemia is present, in which case more frequent monitoring may be indicated. When the affected individual is ill and/or dehydrated, pancreatitis precipitated by hyperlipidemia can be life threatening.
Renal disease
- Urinalysis and measurement of plasma concentrations of electrolytes, uric acid, BUN, and creatinine twice yearly
- Renal and bladder ultrasound examinations every one to two years if the individual is symptomatic or if urinalysis is abnormal
Hepatic disease
- Annual measurement of plasma ALT, AST, and GGT concentration
- Ultrasonography to evaluate for possible steatosis, hepatomegaly, cirrhosis, and portal hypertension
Pulmonary disease. Pulmonary function tests should be performed yearly to evaluate general lung function, even if symptoms of pulmonary fibrosis are not yet present.
Hypothyroidism. Patients should be monitored annually for thyroid abnormalities.
Agents/Circumstances to Avoid
Any substance contraindicated in persons with renal or cardiac failure should be avoided.
Therapy directed at one system may have adverse effects on other systems; for example, the use of glitazone therapy in diabetes mellitus is contraindicated in the presence of cardiac failure.
Evaluation of Relatives at Risk
Early evaluation and/or molecular genetic testing (if the two pathogenic allelic variants in a family are known) of at-risk sibs allows for early diagnosis and early treatment of manifestations.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
There are no data to guide the management of a pregnancy should it occur in a female with Alström syndrome. Potential maternal complications during pregnancy could include development or worsening of cardiac, renal, or hepatic failure; severe dyslipidemia; and hyperglycemia. The fetus could be affected by placental dysfunction resulting from maternal diabetes and cardiorenal disease. Intensive multidisciplinary surveillance of any pregnancy would be mandatory.
Therapies Under Investigation
Search ClinicalTrials.gov for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members. This section is not meant to address all personal, cultural, or ethical issues that individuals may face or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Alström syndrome is inherited in an autosomal recessive manner.
Risk to Family Members
Parents of a proband
- Heterozygotes (carriers) are asymptomatic.
Sibs of a proband
- Once an at-risk sib is known to be unaffected, the chance of his/her being a carrier is 2/3.
Offspring of a proband. No individuals with molecularly confirmed Alström syndrome are known to be fertile.
Other family members of a proband. Each sib of the proband's parents is at a 50% risk of being a carrier.
Carrier Detection
Carrier testing for at-risk family members is possible if the pathogenic variants have been identified in a family member.
Related Genetic Counseling Issues
See Management, Evaluation of Relatives at Risk for information on evaluating at-risk relatives for the purpose of early diagnosis and treatment.
Family planning
- The optimal time for determination of genetic risk, clarification of carrier status, and discussion of the availability of prenatal testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected, are carriers, or are at risk of being carriers.
DNA banking is the storage of DNA (typically extracted from white blood cells) for possible future use. Because it is likely that testing methodology and our understanding of genes, allelic variants, and diseases will improve in the future, consideration should be given to banking DNA of affected individuals.
Prenatal Testing and Preimplantation Genetic Diagnosis
Once the ALMS1 pathogenic variants have been identified in the family, prenatal diagnosis or preimplantation genetic diagnosis for a pregnancy at increased risk for Alström syndrome may be an option that a couple may wish to consider.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- Alstrom Syndrome International (ASI)14 Whitney Farm RoadMount Desert ME 04660Phone: 800-371-3628 (toll-free)Fax: 207-288-6078Email: jdm@jax.org
- Alstrom Syndrome UK Support Group49 Southfield AvenuePaignton Devon TQ3 1LHUnited KingdomPhone: 1803 524238Email: info@alstrom.org.uk
- My46 Trait Profile
- National Library of Medicine Genetics Home Reference
- Ciliopathy AllianceUnited KingdomPhone: 44 20 7387 0543
- Alström Syndrome Registry (ASR)ME
- EURO-WABB Project RegistryAn EU Rare Diseases Registry for Wolfram syndrome, Alstrom syndrome, Bardet-Biedl syndrome and other rare diabetes syndromes.
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Alstrom Syndrome: Genes and Databases
| Gene | Chromosome Locus | Protein | Locus Specific | HGMD |
|---|---|---|---|---|
| ALMS1 | 2p13.1 | Alstrom syndrome protein 1 | Leeds
Mutation Database (ALMS1) ALMS1 database |
ALMS1 |
Table B.
OMIM Entries for Alstrom Syndrome (View All in OMIM)
Gene structure.ALMS1 is a novel gene that does not share significant sequence homology with any other genes. There are 23 exons in ALMS1 (Reference sequence NM_015120.4). For a detailed summary of gene and protein information, see Table A, Gene.
Pathogenic allelic variants. To date, more than 130 pathogenic variants in ALMS1 have been reported in Alström syndrome. The majority of these are nonsense and frameshift variants (insertions or deletions) that are predicted to cause premature protein truncation [Collin et al 2002, Hearn et al 2002, Kinoshita et al 2003, Titomanlio et al 2004, Bond et al 2005, Minton et al 2006, Joy et al 2007, Marshall et al 2007b, Malm et al 2008, Aldahmesh et al 2009, Liu et al 2009, Izzi et al 2011, Kocova et al 2011, Marshall et al 2011a, Marshall et al 2011b, Pereiro et al 2011, Wang et al 2011, Aliferis et al 2012, Taşkesen et al 2012].
Normal gene product. The 12.9 kb ALMS1 transcript encodes a ubiquitously expressed protein of 4,169 amino acids of unknown function 9 (Reference sequence NP_055935.4).
Abnormal gene product. ALMS1 protein localizes to the centrosomes and basal bodies of ciliated cells, suggesting a role for ALMS1 in intracellular trafficking and ciliary function [Collin et al 2005, Hearn et al 2005, Li et al 2007, Jagger et al 2011].
References
Literature Cited
- Aldahmesh MA, Abu-Safieh L, Khan AO, Al-Hassnan ZN, Shaheen R, Rajab M, Monies D, Meyer BF, Alkuraya FS. Allelic heterogeneity in inbred populations: the Saudi experience with Alström syndrome as an illustrative example. Am J Med Genet A. 2009;149A:662–5. [PubMed]
- Aliferis K, Hellé S, Gyapay G, Duchatelet S, Stoetzel C, Mandel JL, Dollfus H. Differentiating Alström from Bardet-Biedl syndrome (BBS) using systematic ciliopathy genes sequencing. Ophthalmic Genet. 2012;33:18–22. [PubMed]
- Bell CJ, Dinwiddie DL, Miller NA, Hateley SL, Ganusova EE, Mudge J, Langley RJ, Zhang L, Lee CC, Schilkey FD, Sheth V, Woodward JE, Peckham HE, Schroth GP, Kim RW, Kingsmore SF. Carrier testing for severe childhood recessive diseases by next-generation sequencing. Sci Transl Med. 2011;3:65ra4. [PMC free article ] [PubMed]
- Bond J, Flintoff K, Higgins J, Scott S, Bennet C, Parsons J, Mannon J, Jafri H, Rashid Y, Barrow M, Trembath R, Woodruff G, Rossa E, Lynch S, Sheilds J, Newbury-Ecob R, Falconer A, Holland P, Cockburn D, Karbani G, Malik S, Ahmed M, Roberts E, Taylor G, Woods CG. The importance of seeking ALMS1 mutations in infants with dilated cardiomyopathy. J Med Genet. 2005;42:e10. [PMC free article ] [PubMed]
- Collin GB, Marshall JD, Ikeda A, So WV, Russell-Eggitt I, Maffei P, Beck S, Boerkoel CF, Sicolo N, Martin M, Nishina PM, Naggert JK. Mutations in ALMS1 cause obesity, type 2 diabetes and neurosensory degeneration in Alstrom syndrome. Nat Genet. 2002;31:74–8. [PubMed]
- Collin GB, Cyr E, Bronson R, Marshall JD, Gifford EJ, Hicks W, Murray SA, Zheng QY, Smith RS, Nishina PM, Naggert JK. Alms1-disrupted mice recapitulate human Alstrom syndrome. Hum Mol Genet. 2005;14:2323–33. [PMC free article ] [PubMed]
- Deeble VJ, Roberts E, Jackson A, Lench N, Karbani G, Woods CG. The continuing failure to recognise Alstrom syndrome and further evidence of genetic homogeneity. J Med Genet. 2000;37:219. [PMC free article ] [PubMed]
- Florentzson R, Hallén K, Möller C. Alström syndrome and cochlear implantation. The first clinical experience. Stockholm, Sweden: 10th International CI Conference. 2010.
- Goerler H, Warnecke G, Winterhalter M, Müller C, Ballmann M, Wessel A, Haverich A, Strüber M, Simon A. Heart-lung transplantation in a 14-year-old boy with Alström syndrome. J Heart Lung Transplant. 2007;26:1217–8. [PubMed]
- Goldstone AP, Beales PL. Genetic obesity syndromes. Front Horm Res. 2008;36:37–60. [PubMed]
- Hearn T, Renforth GL, Spalluto C, Hanley NA, Piper K, Brickwood S, White C, Connolly V, Taylor JF, Russell-Eggitt I, Bonneau D, Walker M, Wilson DI. Mutation of ALMS1, a large gene with a tandem repeat encoding 47 amino acids, causes Alstrom syndrome. Nat Genet. 2002;31:79–83. [PubMed]
- Hearn T, Spalluto C, Phillips VJ, Renforth GL, Copin N, Hanley NA, Wilson DI. Subcellular localization of ALMS1 supports involvement of centrosome and basal body dysfunction in the pathogenesis of obesity, insulin resistance, and type 2 diabetes. Diabetes. 2005;54:1581–7. [PubMed]
- Hoffman JD, Jacobson Z, Young TL, Marshall JD, Kaplan P. Familial variable expression of dilated cardiomyopathy in Alstrom syndrome: a report of four sibs. Am J Med Genet A. 2005;135:96–8. [PubMed]
- Iannello S, Bosco P, Camuto M, Cavaleri A, Milazzo P, Belfiore F. A mild form of Alstrom disease associated with metabolic syndrome and very high fasting serum free fatty acids: two cases diagnosed in adult age. Am J Med Sci. 2004;327:284–8. [PubMed]
- Izzi C, Maffei P, Milan G, Tardanico R, Foini P, Marshall J, Marega A, Scolari F. The Case: Familial occurrence of retinitis pigmentosa, deafness, and nephropathy. Kidney Int. 2011;79:691–2. [PMC free article ] [PubMed]
- Jagger D, Collin G, Kelly J, Towers E, Nevill G, Longo-Guess C, Benson J, Halsey K, Dolan D, Marshall J, Naggert J, Forge A. Alstrom Syndrome protein ALMS1 localizes to basal bodies of cochlear hair cells and regulates cilium-dependent planar cell polarity. Hum Mol Genet. 2011;20:466–81. [PMC free article ] [PubMed]
- Jatti K, Paisey R, More R. Coronary artery disease in Alström syndrome. Eur J Hum Genet. 2012;20:117–8. [PMC free article ] [PubMed]
- Joy T, Cao H, Black G, Malik R, Charlton-Menys V, Hegele RA, Durrington PN. Alstrom syndrome (OMIM 203800): a case report and literature review. Orphanet J Rare Dis. 2007 Dec 21;2:49. [PMC free article ] [PubMed]
- Khoo EY, Risley J, Zaitoun AM, El-Sheikh M, Paisey RB, Acheson AG, Mansell P. Alström syndrome and cecal volvulus in 2 siblings. Am J Med Sci. 2009;337:383–5. [PubMed]
- Kinoshita T, Hanaki K, Kawashima Y, Nagaishi J, Hayashi A, Okada S, Murakami J, Nanba E, Tomonaga R, Kanzaki S. A novel non-sense mutation in Alstrom syndrome: subcellular localization of its truncated protein. Clin Pediatr Endocrinol. 2003;12:114.
- Koç E, Bayrak G, Suher M, Ensari C, Aktas D, Ensari A. Rare case of Alstrom syndrome without obesity and with short stature, diagnosed in adulthood. Nephrology. 2006;11:81–4. [PubMed]
- Kocova M, Sukarova-Angelovska E, Kacarska R, Maffei P, Milan G, Marshall JD. The unique combination of dermatological and ocular phenotypes in Alström syndrome: Severe presentation, early onset, and two novel ALMS1 mutations. Br J Dermatol. 2011;164:878–80. [PMC free article ] [PubMed]
- Koray F, Corter C, Benderli Y, Satman I, Yilmaz T, Dinccag N, Karsidag K. Alström syndrome: a case report. J Oral Sci. 2001;43:221–4. [PubMed]
- Li G, Vega R, Nelms K, Gekakis N, Goodnow C, McNamara P, Wu H, Hong N, Glynne R. A role for Alstrom syndrome protein, Alms1, in kidney ciliogenesis and cellular quiescence. PLoS Genet. 2007;3:e8. [PMC free article ] [PubMed]
- Liu L., Dong B, Chen X, Li J, Li Y. Identification of a novel ALMS1 mutation in a Chinese family with Alström syndrome. Eye. 2009;23:1210–2. [PubMed]
- Loudon MA, Bellenger NG, Carey CM, Paisey RB. Cardiac magnetic resonance imaging in Alström syndrome. Orphanet J Rare Dis. 2009;4:14. [PMC free article ] [PubMed]
- MacDermott S. Urological involvement in Alstrom syndrome. Ottawa, Canada: Alstrom Syndrome International Conference. 2001.
- Maffei P, Boschetti M, Marshall JD, Paisey RB, Beck S, Resmini E, Collin GB, Naggert JK, Milan G, Vettor R, Minuto F, Sicolo N, Barreca A. Characterization of the IGF system in 15 patients with Alström syndrome. Clin Endocrinol (Oxf) 2007;66:269–75. [PubMed]
- Maffei P, Munno V, Marshall JD, Scandellari C, Sicolo N. The Alstrom syndrome: is it a rare or unknown disease? Ann Ital Med Int. 2002;17:221–8. [PubMed]
- Makaryus AN, Popowski B, Kort S, Paris Y, Mangion J. A rare case of Alström syndrome presenting with rapidly progressive severe dilated cardiomyopathy diagnosed by echocardiography. J Am Soc Echocardiogr. 2003;16:194–6. [PubMed]
- Malm E, Ponjavic V, Nishina PM, Naggert JK, Hinman EG, Andréasson S, Marshall JD, Möller C. Full-field electroretinography and marked variability in clinical phenotype of Alström syndrome. Arch Ophthalmol. 2008;126:51–7. [PubMed]
- Marshall JD, Bronson RT, Collin GB, Nordstrom AD, Maffei P, Paisey RB, Carey C, Macdermott S, Russell-Eggitt I, Shea SE, Davis J, Beck S, Shatirishvili G, Mihai CM, Hoeltzenbein M, Pozzan GB, Hopkinson I, Sicolo N, Naggert JK, Nishina PM. New Alstrom syndrome phenotypes based on the evaluation of 182 cases. Arch Intern Med. 2005;165:675–83. [PubMed]
- Marshall JD, Beck S, Maffei P, Naggert JK. Alström Syndrome. Eur J Hum Genet. 2007a;15:1193–202. [PubMed]
- Marshall JD, Hinman EG, Collin GB, Beck S, Cerqueira R, Maffei P, Milan G, Zhang W, Wilson DI, Hearn T, Tavares P, Vettor R, Veronese C, Martin M, So WV, Nishina PM, Naggert JK. Spectrum of ALMS1 variants and evaluation of genotype-phenotype correlations in Alström syndrome. Hum Mutat. 2007b;28:1114–23. [PubMed]
- Marshall JD, Maffei P, Beck S, Barrett TG, Paisey RB. Clinical utility gene card for: Alström syndrome. Eur J Hum Genet. 2011a;19 [PMC free article ] [PubMed] [Cross Ref]
- Marshall JD, Maffei P, Collin GB, Naggert JK. Alstrom Syndrome: Genetics and Clinical Overview. Curr Genomics. 2011b;12:225–35. [PMC free article ] [PubMed]
- Minton JA, Owen KR, Ricketts CJ, Crabtree N, Shaikh G, Ehtisham S, Porter JR, Carey C, Hodge D, Paisey R, Walker M, Barrett TG. Syndromic obesity and diabetes: changes in body composition with age and mutation analysis of ALMS1 in 12 United Kingdom kindreds with Alstrom syndrome. J Clin Endocrinol Metab. 2006;91:3110–6. [PubMed]
- Ozgül RK, Satman I, Collin GB, Hinman EG, Marshall JD, Kocaman O, Tütüncü Y, Yilmaz T, Naggert JK. Molecular analysis and long-term clinical evaluation of three siblings with Alström syndrome. Clin Genet. 2007;72:351–6. [PubMed]
- Paisey RB, Carey CM, Bower L, Marshall J, Taylor P, Maffei P, Mansell P. Hypertriglyceridaemia in Alström's syndrome: causes and associations in 37 cases. Clin Endocrinol (Oxf) 2004;2004;60:228–31. [PubMed]
- Paisey RB. New insights and therapies for the metabolic consequences of Alström syndrome. Curr Opin Lipidol. 2009;20:315–20. [PubMed]
- Paisey RB, Hodge D, Bower L. Weight and glycaemic responses to 6 months Exanetide treatment in 9 Alstrom syndrome subjects with type 2 diabetes. Abstract 288. Rome, Italy: 44th Meeting of the European Association for the Study of Diabetes. 2008.
- Paisey RB, Paisey RM, Thomson MP, Bower L, Maffei P, Shield JP, Barnett S, Marshall JD. Protection from clinical peripheral sensory neuropathy in Alström syndrome in contrast to early-onset type 2 diabetes. Diabetes Care. 2009;32:462–4. [PMC free article ] [PubMed]
- Pereiro I, Hoskins BE, Marshall JD, Collin GB, Naggert JK, Piñeiro-Gallego T, Oitmaa E, Katsanis N, Valverde D, Beales PL. Arrayed Primer Extension (APEX) technology simplifies mutation detection in Bardet Biedl and Alström Syndrome. Eur J Hum Genet. 2011;19:485–8. [PMC free article ] [PubMed]
- Quiros-Tejeira RE, Vargas J, Ament ME. Early-onset liver disease complicated with acute liver failure in Alstrom syndrome. Am J Med Genet. 2001;101:9–11. [PubMed]
- Taşkesen M, Collin GB, Evsikov AV, Güzel A, Ozgül RK, Marshall JD, Naggert JK. Novel Alu retrotransposon insertion leading to Alström syndrome. Hum Genet. 2012;131:407–13. [PMC free article ] [PubMed]
- Titomanlio L, De Brasi D, Buoninconti A, Sperandeo MP, Pepe A, Andria G, Sebastio G. Alstrom syndrome: intrafamilial phenotypic variability in sibs with a novel nonsense mutation of the ALMS1 gene. Clin Genet. 2004;65:156–7. [PubMed]
- Van den Abeele K, Craen M, Schuil J, Meire FM. Ophthalmologic and systemic features of the Alström syndrome: report of 9 cases. Bull Soc Belge Ophtalmol. 2001;281:67–72. [PubMed]
- Wang X, Wang H, Cao M, Li Z, Chen X, Patenia C, Gore A, Abboud EB, Al-Rajhi AA. A Lewis R, Lupski JR, Mardon G, Zhang K, Muzny D, Gibbs RA, Chen R. Whole-exome sequencing identifies ALMS1, IQCB1, CNGA3, and MYO7A mutations in patients with Leber congenital amaurosis. Hum Mutat. 2011;32:1450–9. [PMC free article ] [PubMed]
- Wu WC, Chen SC, Dia CY, Yu ML, Hsieh MY, Lin ZY, Wang LY, Tsai JF, Chang WY, Chuang WL. Alstrom syndrome with acute pancreatitis: a case report. Kaohsiung J Med Sci. 2003;19:358–61. [PubMed]
Chapter Notes
Author History
Catherine Carey, MD, FRCP (2003-present)
Ian Hopkinson, BSc
(Hons), MBChB, PhD, MRCGP; University College London (2003-2010)
Seamus Macdermott, MD, FRCS (2003-present)
Jan D Marshall, MS
(2003-present)
Richard B Paisey, MD, FRCP (2005-present)
Revision History
- 31 May 2012 (me) Comprehensive update posted live
- 8 June 2010 (me) Comprehensive update posted live
- 25 June 2007 (me) Comprehensive update posted to live Web site
- 7 February 2005 (me) Comprehensive update posted to live Web site
- 11 May 2004 (ih) Revision: test availability
- 7 February 2003 (me) Review posted to live Web site
- 6 June 2002 (ih) Original submission